当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A molecular evolution algorithm for ligand design in DOCK
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-09-08 , DOI: 10.1002/jcc.26993 Lauren E Prentis 1 , Courtney D Singleton 2 , John D Bickel 3 , William J Allen 4 , Robert C Rizzo 4, 5, 6
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-09-08 , DOI: 10.1002/jcc.26993 Lauren E Prentis 1 , Courtney D Singleton 2 , John D Bickel 3 , William J Allen 4 , Robert C Rizzo 4, 5, 6
Affiliation
|
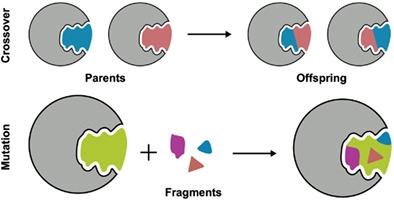
As a complement to virtual screening, de novo design of small molecules is an alternative approach for identifying potential drug candidates. Here, we present a new 3D genetic algorithm to evolve molecules through breeding, mutation, fitness pressure, and selection. The method, termed DOCK_GA, builds upon and leverages powerful sampling, scoring, and searching routines previously implemented into DOCK6. Three primary experiments were used during development: Single-molecule evolution evaluated three selection methods (elitism, tournament, and roulette), in four clinically relevant systems, in terms of mutation type and crossover success, chemical properties, ensemble diversity, and fitness convergence, among others. Large scale benchmarking assessed performance across 651 different protein-ligand systems. Ensemble-based evolution demonstrated using multiple inhibitors simultaneously to seed growth in a SARS-CoV-2 target. Key takeaways include: (1) The algorithm is robust as demonstrated by the successful evolution of molecules across a large diverse dataset. (2) Users have flexibility with regards to parent input, selection method, fitness function, and molecular descriptors. (3) The program is straightforward to run and only requires a single executable and input file at run-time. (4) The elitism selection method yields more tightly clustered molecules in terms of 2D/3D similarity, with more favorable fitness, followed by tournament and roulette.
中文翻译:

DOCK 中配体设计的分子进化算法
作为虚拟筛选的补充,小分子的从头设计是识别潜在候选药物的另一种方法。在这里,我们提出了一种新的 3D 遗传算法,通过育种、突变、适应压力和选择来进化分子。该方法称为 DOCK_GA,建立并利用了先前在 DOCK6 中实现的强大采样、评分和搜索例程。开发过程中使用了三个主要实验:单分子进化在四个临床相关系统中评估了三种选择方法(精英主义、锦标赛和轮盘赌),包括突变类型和交叉成功、化学性质、整体多样性和适应度收敛性。除其他外。大规模基准测试评估了 651 种不同蛋白质配体系统的性能。基于整体的进化证明了同时使用多种抑制剂在 SARS-CoV-2 靶标中促进生长。主要结论包括:(1)该算法是稳健的,分子在大型多样化数据集中的成功进化证明了这一点。 (2) 用户在亲本输入、选择方法、适应度函数和分子描述符方面具有灵活性。 (3) 该程序运行起来很简单,运行时只需要一个可执行文件和输入文件。 (4)精英选择方法在2D/3D相似性方面产生更紧密聚集的分子,具有更有利的适应度,其次是锦标赛和轮盘赌。
更新日期:2022-09-08
中文翻译:
DOCK 中配体设计的分子进化算法
作为虚拟筛选的补充,小分子的从头设计是识别潜在候选药物的另一种方法。在这里,我们提出了一种新的 3D 遗传算法,通过育种、突变、适应压力和选择来进化分子。该方法称为 DOCK_GA,建立并利用了先前在 DOCK6 中实现的强大采样、评分和搜索例程。开发过程中使用了三个主要实验:单分子进化在四个临床相关系统中评估了三种选择方法(精英主义、锦标赛和轮盘赌),包括突变类型和交叉成功、化学性质、整体多样性和适应度收敛性。除其他外。大规模基准测试评估了 651 种不同蛋白质配体系统的性能。基于整体的进化证明了同时使用多种抑制剂在 SARS-CoV-2 靶标中促进生长。主要结论包括:(1)该算法是稳健的,分子在大型多样化数据集中的成功进化证明了这一点。 (2) 用户在亲本输入、选择方法、适应度函数和分子描述符方面具有灵活性。 (3) 该程序运行起来很简单,运行时只需要一个可执行文件和输入文件。 (4)精英选择方法在2D/3D相似性方面产生更紧密聚集的分子,具有更有利的适应度,其次是锦标赛和轮盘赌。










































 京公网安备 11010802027423号
京公网安备 11010802027423号