当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electrochemical CO2 Reduction over Metal-/Nitrogen-Doped Graphene Single-Atom Catalysts Modeled Using the Grand-Canonical Density Functional Theory
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-08-04 , DOI: 10.1021/acscatal.2c01832 Paige Brimley 1 , Hussain Almajed 1 , Yousef Alsunni 1, 2 , Abdulaziz W. Alherz 1, 3 , Zachary J. L. Bare 1 , Wilson A. Smith 1, 4, 5, 6 , Charles B. Musgrave 1, 4, 7
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-08-04 , DOI: 10.1021/acscatal.2c01832 Paige Brimley 1 , Hussain Almajed 1 , Yousef Alsunni 1, 2 , Abdulaziz W. Alherz 1, 3 , Zachary J. L. Bare 1 , Wilson A. Smith 1, 4, 5, 6 , Charles B. Musgrave 1, 4, 7
Affiliation
|
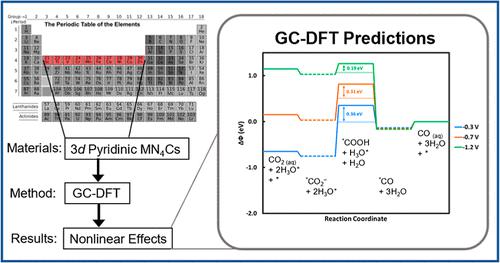
Renewably driven, electrochemical conversion of carbon dioxide into value-added products is expected to be a critical tool in global decarbonization. However, theoretical studies based on the computational hydrogen electrode largely ignore the nonlinear effects of the applied potential on the calculated results, leading to inaccurate predictions of catalytic behavior or mechanistic pathways. Here, we use grand canonical density functional theory (GC-DFT) to model electrochemical CO2 reduction (CO2R) over metal- and nitrogen-doped graphene catalysts (MNCs) and explicitly include the effects of the applied potential. We used GC-DFT to compute the CO2 to CO reaction intermediate energies at −0.3, −0.7, and −1.2 VSHE catalyzed by MNCs each doped with 1 of the 10 3d block metals coordinated by four pyridinic nitrogen atoms. Our results predict that Sc-, Ti-, Co-, Cu-, and Zn-N4Cs effectively catalyze CO2R at moderate to large reducing potentials (−0.7 to −1.2 VSHE). ZnN4C is a particularly promising electrocatalyst for CO2R to CO both at low and moderate applied potentials based on our thermodynamic analysis. Our findings also explain the observed pH independence of CO production over FeN4C and predict that the rate-determining step of CO2R over FeN4C is not *CO2– formation but rather *CO desorption. Additionally, the GC-DFT-computed density of states analysis illustrates how the electronic states of MNCs and adsorbates change non-uniformly with applied potential, resulting in a significantly increased *CO2– stability relative to other intermediates and demonstrating that the formation of the adsorbed *CO2– anion is critical to CO2R activation. This work demonstrates how GC-DFT paves the way for physically realistic and accurate theoretical simulations of reacting electrochemical systems.
中文翻译:

使用大规范密度泛函理论模拟的金属/氮掺杂石墨烯单原子催化剂上的电化学 CO2 还原
可再生能源驱动,将二氧化碳电化学转化为增值产品预计将成为全球脱碳的关键工具。然而,基于计算氢电极的理论研究在很大程度上忽略了施加电位对计算结果的非线性影响,导致对催化行为或机理途径的预测不准确。在这里,我们使用大规范密度泛函理论 (GC-DFT) 来模拟金属和氮掺杂石墨烯催化剂 (MNC) 上的电化学 CO 2还原 (CO 2 R),并明确包括应用电位的影响。我们使用 GC-DFT 计算了在 -0.3、-0.7 和 -1.2 V SHE下的 CO 2到 CO 反应中间体能量由 MNC 催化,每个掺杂有 10 个 3d 嵌段金属中的 1 个,由四个吡啶氮原子配位。我们的结果预测,Sc-、Ti-、Co-、Cu- 和 Zn-N 4 Cs在中等到大的还原电位(-0.7 到 -1.2 V SHE)下有效地催化 CO 2 R。根据我们的热力学分析,ZnN 4 C 是一种特别有前途的用于 CO 2 R 到 CO 的电催化剂,无论是在低应用电位还是中等应用电位下。我们的研究结果还解释了在 FeN 4 C上观察到的 CO 产生的 pH 独立性,并预测 CO 2 R 在 FeN 4 C 上的速率决定步骤不是 *CO 2 –形成而是*CO解吸。此外,GC-DFT 计算的状态密度分析说明了 MNC 和吸附物的电子状态如何随施加的电位不均匀地变化,导致 *CO 2 -相对于其他中间体的稳定性显着增加,并证明形成吸附的*CO 2 –阴离子对CO 2 R 活化至关重要。这项工作展示了 GC-DFT 如何为反应电化学系统的物理现实和准确的理论模拟铺平道路。
更新日期:2022-08-04
中文翻译:
使用大规范密度泛函理论模拟的金属/氮掺杂石墨烯单原子催化剂上的电化学 CO2 还原
可再生能源驱动,将二氧化碳电化学转化为增值产品预计将成为全球脱碳的关键工具。然而,基于计算氢电极的理论研究在很大程度上忽略了施加电位对计算结果的非线性影响,导致对催化行为或机理途径的预测不准确。在这里,我们使用大规范密度泛函理论 (GC-DFT) 来模拟金属和氮掺杂石墨烯催化剂 (MNC) 上的电化学 CO 2还原 (CO 2 R),并明确包括应用电位的影响。我们使用 GC-DFT 计算了在 -0.3、-0.7 和 -1.2 V SHE下的 CO 2到 CO 反应中间体能量由 MNC 催化,每个掺杂有 10 个 3d 嵌段金属中的 1 个,由四个吡啶氮原子配位。我们的结果预测,Sc-、Ti-、Co-、Cu- 和 Zn-N 4 Cs在中等到大的还原电位(-0.7 到 -1.2 V SHE)下有效地催化 CO 2 R。根据我们的热力学分析,ZnN 4 C 是一种特别有前途的用于 CO 2 R 到 CO 的电催化剂,无论是在低应用电位还是中等应用电位下。我们的研究结果还解释了在 FeN 4 C上观察到的 CO 产生的 pH 独立性,并预测 CO 2 R 在 FeN 4 C 上的速率决定步骤不是 *CO 2 –形成而是*CO解吸。此外,GC-DFT 计算的状态密度分析说明了 MNC 和吸附物的电子状态如何随施加的电位不均匀地变化,导致 *CO 2 -相对于其他中间体的稳定性显着增加,并证明形成吸附的*CO 2 –阴离子对CO 2 R 活化至关重要。这项工作展示了 GC-DFT 如何为反应电化学系统的物理现实和准确的理论模拟铺平道路。










































 京公网安备 11010802027423号
京公网安备 11010802027423号