Nature Reviews Endocrinology ( IF 31.0 ) Pub Date : 2022-08-03 , DOI: 10.1038/s41574-022-00718-y Isadora P Cavalcante 1 , Annabel Berthon 1 , Maria C Fragoso 2 , Martin Reincke 3 , Constantine A Stratakis 4 , Bruno Ragazzon 1 , Jérôme Bertherat 5
|
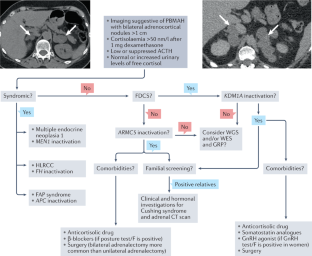
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is an adrenal cause of Cushing syndrome. Nowadays, a PBMAH diagnosis is more frequent than previously, as a result of progress in the diagnostic methods for adrenal incidentalomas, which are widely available. Although some rare syndromic forms of PBMAH are known to be of genetic origin, non-syndromic forms of PBMAH have only been recognized as a genetic disease in the past 10 years. Genomics studies have highlighted the molecular heterogeneity of PBMAH and identified molecular subgroups, allowing improved understanding of the clinical heterogeneity of this disease. Furthermore, the generation of these subgroups permitted the identification of new genes responsible for PBMAH. Constitutive inactivating variants in ARMC5 and KDM1A are responsible for the development of distinct forms of PBMAH. To date, pathogenic variants of ARMC5 are responsible for 20–25% of PBMAH, whereas germline KDM1A alterations have been identified in >90% of PBMAH causing food-dependent Cushing syndrome. The identification of pathogenic variants in ARMC5 and KDM1A demonstrated that PBMAH, despite mostly being diagnosed in adults aged 45–60 years, is a genetic disorder. This Review summarizes the important progress made in the past 10 years in understanding the genetics of PBMAH, which have led to a better understanding of the pathophysiology, opening new clinical perspectives.
中文翻译:
原发性双侧大结节性肾上腺增生:绝对是遗传病
原发性双侧大结节性肾上腺增生 (PBMAH) 是库欣综合征的肾上腺原因。如今,由于广泛使用的肾上腺偶发瘤诊断方法的进步,PBMAH 的诊断比以前更频繁。尽管已知一些罕见的综合征形式的 PBMAH 具有遗传起源,但非综合征形式的 PBMAH 仅在过去 10 年中才被认为是一种遗传疾病。基因组学研究强调了 PBMAH 的分子异质性并确定了分子亚群,从而提高了对该疾病临床异质性的理解。此外,这些亚组的产生允许鉴定负责PBMAH的新基因。ARMC5和KDM1A中的组成型失活变体负责开发不同形式的PBMAH。迄今为止,ARMC5 的致病性变异是导致20-25% 的 PBMAH 的原因,而在超过 90% 的 PBMAH 中发现了种系KDM1A改变,导致食物依赖性库欣综合征。对ARMC5和KDM1A致病变异的鉴定表明,PBMAH 尽管主要在 45-60 岁的成年人中被诊断出来,但它是一种遗传性疾病。本综述总结了过去 10 年在了解 PBMAH 遗传学方面取得的重要进展,这些进展导致了对病理生理学的更好理解,开辟了新的临床视角。










































 京公网安备 11010802027423号
京公网安备 11010802027423号