当前位置:
X-MOL 学术
›
Am. J. Hematol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The pleiotropic effects of α-thalassemia on HbSS and HbSC sickle cell disease: Reduced erythrocyte cation co-transport activity, serum erythropoietin, and transfusion burden, do not translate into increased survival
American Journal of Hematology ( IF 10.1 ) Pub Date : 2022-07-08 , DOI: 10.1002/ajh.26652 John N Brewin 1, 2 , Amina Nardo-Marino 1, 2, 3 , Sara Stuart-Smith 1 , Sara El Hoss 2 , Anke Hanneman 4 , John Strouboulis 2 , Stephan Menzel 2 , John S Gibson 4 , David C Rees 1, 2
American Journal of Hematology ( IF 10.1 ) Pub Date : 2022-07-08 , DOI: 10.1002/ajh.26652 John N Brewin 1, 2 , Amina Nardo-Marino 1, 2, 3 , Sara Stuart-Smith 1 , Sara El Hoss 2 , Anke Hanneman 4 , John Strouboulis 2 , Stephan Menzel 2 , John S Gibson 4 , David C Rees 1, 2
Affiliation
|
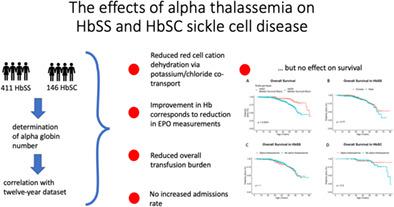
α-Thalassemia is one of the most important genetic modulators of sickle cell disease (SCD). Both beneficial and detrimental effects have been described previously. We use a 12-year data set on a large cohort of patients with HbSS (n = 411) and HbSC (n = 146) to examine a wide range of these clinical and laboratory associations. Our novel findings are that α-thalassemia strongly reduces erythrocyte potassium chloride co-transporter (KCC) activity in both HbSS and HbSC (p = .035 and p = .00045 respectively), suggesting a novel mechanism through which α-thalassemia induces a milder phenotype by reducing red cell cation loss. This may be particularly important in HbSC where reduction in mean cell hemoglobin concentration is not seen and where KCC activity has previously been found to correlate with disease severity. Additionally, we show that α-thalassemia not only increases hemoglobin in patients with HbSS (p = .0009) but also reduces erythropoietin values (p = .0005), demonstrating a measurable response to improved tissue oxygenation. We confirm the reno-protective effect of α-thalassemia in patients with HbSS, with reduced proteinuria (p = .003) and demonstrate a novel association with increased serum sodium (p = .0004) and reduced serum potassium values (p = 5.74 × 10−10). We found patients with α-thalassemia had a reduced annualized transfusion burden in both HbSS and HbSC, but α-thalassemia had no impact on annualized admission rates in either group. Finally, in a larger cohort, we report a median survival of 62 years in patients with HbSS (n = 899) and 80 years in those with HbSC (n = 240). α-thalassemia did not influence survival in HbSS, but a nonsignificant trend was seen in those with HbSC.
中文翻译:

α-地中海贫血对 HbSS 和 HbSC 镰状细胞病的多效性影响:减少红细胞阳离子共转运活性、血清促红细胞生成素和输血负担,不会转化为增加的存活率
α-地中海贫血是镰状细胞病 (SCD) 最重要的遗传调节剂之一。前面已经描述了有益和有害的影响。我们使用针对大量 HbSS ( n = 411) 和 HbSC ( n = 146)患者的 12 年数据集来 检查这些临床和实验室关联的广泛范围。我们的新发现是 α-地中海贫血强烈降低 HbSS 和 HbSC 中的红细胞氯化钾协同转运蛋白 (KCC) 活性 ( p = .035 和p = .00045),提出了一种新的机制,α-地中海贫血通过减少红细胞阳离子丢失来诱导更温和的表型。这在 HbSC 中可能特别重要,在 HbSC 中未观察到平均细胞血红蛋白浓度降低,并且之前已发现 KCC 活性与疾病严重程度相关。此外,我们表明 α-地中海贫血不仅会增加 HbSS 患者的血红蛋白 ( p = .0009),还会降低促红细胞生成素值 ( p = .0005),表明对改善的组织氧合有可测量的反应。我们证实了 α-地中海贫血对 HbSS 患者的肾脏保护作用,蛋白尿减少 ( p = .003),并证明了与血清钠增加的新关联 ( p = .0004) 和降低的血清钾值 ( p = 5.74 × 10 -10 )。我们发现 α-地中海贫血患者的 HbSS 和 HbSC 年化输血负担均有所降低,但 α-地中海贫血对两组的年化入院率都没有影响。最后,在更大的队列中,我们报告了 HbSS 患者 ( n = 899) 和 HbSC 患者 ( n = 240)的中位生存期分别为 62 年和 80 年。α-地中海贫血不影响 HbSS 患者的生存,但在 HbSC 患者中观察到不显着的趋势。
更新日期:2022-07-08
中文翻译:
α-地中海贫血对 HbSS 和 HbSC 镰状细胞病的多效性影响:减少红细胞阳离子共转运活性、血清促红细胞生成素和输血负担,不会转化为增加的存活率
α-地中海贫血是镰状细胞病 (SCD) 最重要的遗传调节剂之一。前面已经描述了有益和有害的影响。我们使用针对大量 HbSS ( n = 411) 和 HbSC ( n = 146)患者的 12 年数据集来 检查这些临床和实验室关联的广泛范围。我们的新发现是 α-地中海贫血强烈降低 HbSS 和 HbSC 中的红细胞氯化钾协同转运蛋白 (KCC) 活性 ( p = .035 和p = .00045),提出了一种新的机制,α-地中海贫血通过减少红细胞阳离子丢失来诱导更温和的表型。这在 HbSC 中可能特别重要,在 HbSC 中未观察到平均细胞血红蛋白浓度降低,并且之前已发现 KCC 活性与疾病严重程度相关。此外,我们表明 α-地中海贫血不仅会增加 HbSS 患者的血红蛋白 ( p = .0009),还会降低促红细胞生成素值 ( p = .0005),表明对改善的组织氧合有可测量的反应。我们证实了 α-地中海贫血对 HbSS 患者的肾脏保护作用,蛋白尿减少 ( p = .003),并证明了与血清钠增加的新关联 ( p = .0004) 和降低的血清钾值 ( p = 5.74 × 10 -10 )。我们发现 α-地中海贫血患者的 HbSS 和 HbSC 年化输血负担均有所降低,但 α-地中海贫血对两组的年化入院率都没有影响。最后,在更大的队列中,我们报告了 HbSS 患者 ( n = 899) 和 HbSC 患者 ( n = 240)的中位生存期分别为 62 年和 80 年。α-地中海贫血不影响 HbSS 患者的生存,但在 HbSC 患者中观察到不显着的趋势。










































 京公网安备 11010802027423号
京公网安备 11010802027423号