当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Dynamic Docking of Macrocycles in Bound and Unbound Protein Structures with DynaDock
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-07-07 , DOI: 10.1021/acs.jcim.2c00436 Maximilian Meixner 1 , Martin Zachmann 1 , Sebastian Metzler 1 , Jonathan Scheerer 1 , Martin Zacharias 2 , Iris Antes 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-07-07 , DOI: 10.1021/acs.jcim.2c00436 Maximilian Meixner 1 , Martin Zachmann 1 , Sebastian Metzler 1 , Jonathan Scheerer 1 , Martin Zacharias 2 , Iris Antes 1
Affiliation
|
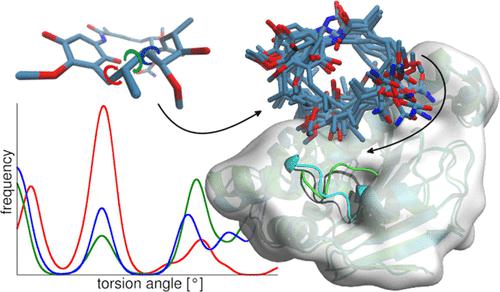
Macrocycles are interesting molecules with unique features due to their conformationally constrained yet flexible ring structure. This characteristic poses a difficult challenge for computational modeling studies since they rely on accurate structural descriptions. In particular, molecular docking calculations suffer from the lack of ring flexibility during pose generation, which is often compensated by using pregenerated ligand conformer ensembles. Moreover, receptor structures are mainly treated rigidly, which limits the use of many docking tools. In this study, we optimized our previous molecular dynamics-based sampling and docking pipeline specifically designed for the accurate prediction of macrocyclic compounds. We developed a dihedral classification procedure for in-depth conformational analysis of the macrocyclic rings and extracted structural ensembles that were subsequently docked in both bound and unbound protein structures employing a fully flexible approach. Our results suggest that including a ring conformer close to the bound state in the starting ensemble increases the chance of successful docking. The bioactive conformations of a diverse set of ligands could be predicted with high and decent accuracy in bound and unbound protein structures, respectively, due to the incorporation of full molecular flexibility in our approach. The remaining unsuccessful docking calculations were mainly caused by large flexible substituents that bind to surface-exposed binding sites, rather than the macrocyclic ring per se and could be further improved by explicit molecular dynamics simulations of the docked complex.
中文翻译:

使用 DynaDock 动态对接结合和未结合蛋白质结构中的大环
大环是有趣的分子,由于其构象受限但灵活的环结构,具有独特的特征。这一特性对计算建模研究提出了艰巨的挑战,因为它们依赖于准确的结构描述。特别是,分子对接计算在姿势生成期间缺乏环灵活性,这通常通过使用预生成的配体构象体集合来补偿。此外,受体结构主要被严格处理,这限制了许多对接工具的使用。在这项研究中,我们优化了我们之前专门为准确预测大环化合物而设计的基于分子动力学的采样和对接管道。我们开发了一种二面体分类程序,用于对大环进行深入的构象分析,并提取结构整体,随后采用完全灵活的方法将其停靠在结合和未结合的蛋白质结构中。我们的结果表明,在起始集合中包含一个接近束缚状态的环构象会增加成功对接的机会。由于在我们的方法中结合了完全的分子灵活性,可以分别在结合和未结合的蛋白质结构中以很高和相当的准确度预测各种配体的生物活性构象。其余不成功的对接计算主要是由与表面暴露的结合位点结合的大型柔性取代基引起的,
更新日期:2022-07-07
中文翻译:
使用 DynaDock 动态对接结合和未结合蛋白质结构中的大环
大环是有趣的分子,由于其构象受限但灵活的环结构,具有独特的特征。这一特性对计算建模研究提出了艰巨的挑战,因为它们依赖于准确的结构描述。特别是,分子对接计算在姿势生成期间缺乏环灵活性,这通常通过使用预生成的配体构象体集合来补偿。此外,受体结构主要被严格处理,这限制了许多对接工具的使用。在这项研究中,我们优化了我们之前专门为准确预测大环化合物而设计的基于分子动力学的采样和对接管道。我们开发了一种二面体分类程序,用于对大环进行深入的构象分析,并提取结构整体,随后采用完全灵活的方法将其停靠在结合和未结合的蛋白质结构中。我们的结果表明,在起始集合中包含一个接近束缚状态的环构象会增加成功对接的机会。由于在我们的方法中结合了完全的分子灵活性,可以分别在结合和未结合的蛋白质结构中以很高和相当的准确度预测各种配体的生物活性构象。其余不成功的对接计算主要是由与表面暴露的结合位点结合的大型柔性取代基引起的,










































 京公网安备 11010802027423号
京公网安备 11010802027423号