Nature Biotechnology ( IF 33.1 ) Pub Date : 2022-06-27 , DOI: 10.1038/s41587-022-01361-8 Fredrik Salmen 1, 2 , Joachim De Jonghe 3, 4 , Tomasz S Kaminski 3, 5 , Anna Alemany 1, 2 , Guillermo E Parada 6 , Joe Verity-Legg 1, 2 , Ayaka Yanagida 7 , Timo N Kohler 3, 8 , Nicholas Battich 1, 2 , Floris van den Brekel 1, 2 , Anna L Ellermann 3 , Alfonso Martinez Arias 9 , Jennifer Nichols 8, 10 , Martin Hemberg 6, 11 , Florian Hollfelder 3 , Alexander van Oudenaarden 1, 2
|
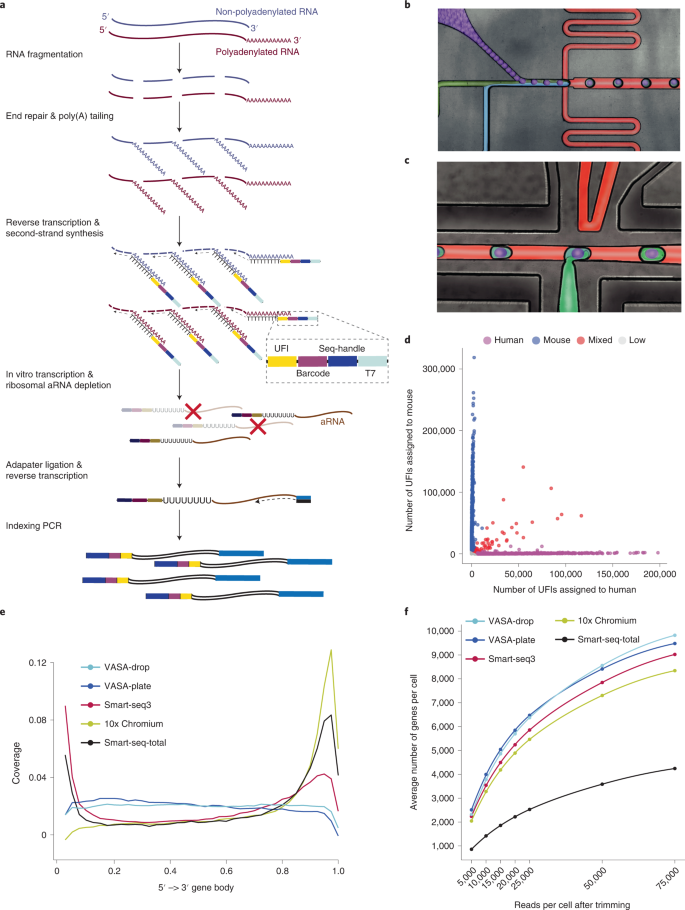
Most methods for single-cell transcriptome sequencing amplify the termini of polyadenylated transcripts, capturing only a small fraction of the total cellular transcriptome. This precludes the detection of many long non-coding, short non-coding and non-polyadenylated protein-coding transcripts and hinders alternative splicing analysis. We, therefore, developed VASA-seq to detect the total transcriptome in single cells, which is enabled by fragmenting and tailing all RNA molecules subsequent to cell lysis. The method is compatible with both plate-based formats and droplet microfluidics. We applied VASA-seq to more than 30,000 single cells in the developing mouse embryo during gastrulation and early organogenesis. Analyzing the dynamics of the total single-cell transcriptome, we discovered cell type markers, many based on non-coding RNA, and performed in vivo cell cycle analysis via detection of non-polyadenylated histone genes. RNA velocity characterization was improved, accurately retracing blood maturation trajectories. Moreover, our VASA-seq data provide a comprehensive analysis of alternative splicing during mammalian development, which highlighted substantial rearrangements during blood development and heart morphogenesis.
中文翻译:
使用 VASA-seq 在单细胞中进行高通量总 RNA 测序
大多数单细胞转录组测序方法都会扩增聚腺苷酸化转录本的末端,仅捕获总细胞转录组的一小部分。这妨碍了许多长非编码、短非编码和非聚腺苷酸化蛋白质编码转录本的检测,并阻碍了选择性剪接分析。因此,我们开发了 VASA-seq 来检测单细胞中的总转录组,这是通过细胞裂解后对所有 RNA 分子进行片段化和加尾来实现的。该方法与基于板的形式和液滴微流体兼容。我们将 VASA-seq 应用到了原肠胚形成和早期器官发生期间发育中的小鼠胚胎中的 30,000 多个单细胞。通过分析总单细胞转录组的动态,我们发现了细胞类型标记,其中许多基于非编码 RNA,并通过检测非聚腺苷酸化组蛋白基因进行体内细胞周期分析。 RNA 速度表征得到改进,可以准确地追踪血液成熟轨迹。此外,我们的 VASA-seq 数据提供了对哺乳动物发育过程中选择性剪接的全面分析,突出了血液发育和心脏形态发生过程中的大量重排。










































 京公网安备 11010802027423号
京公网安备 11010802027423号