Advanced Drug Delivery Reviews ( IF 15.2 ) Pub Date : 2022-06-25 , DOI: 10.1016/j.addr.2022.114402 Mia Horowitz 1 , Hila Braunstein 1 , Ari Zimran 2 , Shoshana Revel-Vilk 2 , Ozlem Goker-Alpan 3
|
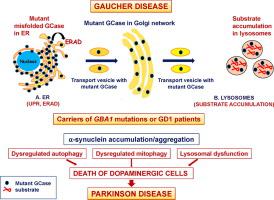
Lysosomes have a critical role in maintaining normal cellular homeostasis mediated by their involvement in secretion, plasma membrane repair, cell signaling and energy metabolism. Lysosomal storage disorders (LSDs) are a group of approximately 50 rare disorders caused by lysosomal dysfunction that occur due to mutations in a gene of a lysosomal protein. Gaucher disease (GD), an autosomal recessive disorder and one of the most common LSDs, is caused by the deficiency of the lysosomal enzyme acid-β-glucocerebrosidase (GCase), due to biallelic mutations in the GBA1 gene. Reduced GCase activity leads to the accumulation of glucosylceramide (GlcCer), which is deacylated by lysosomal acid ceramidase to a toxic metabolite, glucosylshpingosine (GlcSph). Most GBA1 variants are recognized as misfolded in the ER, where the retention for refolding attempts initiates stress and activates the stress response known as the Unfolded Protein Response (UPR).
The distinct clinical subtypes of GD are based on whether there is primary involvement of the central nervous system. Type 1 GD (GD1) is the nonneuropathic type, however, the recent recognition of the association of GD with the development of parkinsonism defies this classification. Patients with GD1 and carriers of GBA1 mutations are at risk for the development of parkinsonian manifestations. Parkinson disease (PD), the second most prevalent neurodegenerative disease, culminates in a movement disorder with the premature death of the patients. In PD and related disorders, collectively called synucleinopathies, the hallmark pathology is α-synuclein positive aggregates referred to as Lewy bodies or Lewy neurites and the death of dopaminergic neurons. While PD is mostly sporadic, in ∼5–10% of cases, the disease results from pathogenic variants in a growing number of genes. The most common genetic cause of PD is mutations in GBA1. Two mechanisms have been proposed for this link: (A) a “gain of function” mechanism, in which mutant GCase (protein) contributes to aggregate formation and to the development of PD, and the (B) “haploinsufficiency” (“loss of function”) model, suggesting that one normal GBA1 allele is insufficient to carry adequate GCase activity and functional deficiency of GCase impedes α-synuclein metabolism. Lysosomal dysfunction, compromised autophagy and mitophagy further enhance the accumulation of α-synuclein, which results in the development of PD pathology.
The present review will elaborate on the biology of GD, its association with PD and related disorders, and discuss the possible mechanisms underlying this association.
中文翻译:
溶酶体功能和功能障碍:戈谢病的分子和细胞机制及其与帕金森病的关联
溶酶体通过参与分泌、质膜修复、细胞信号传导和能量代谢介导,在维持正常细胞稳态方面具有关键作用。溶酶体贮积症 (LSD) 是一组大约 50 种由溶酶体功能障碍引起的罕见疾病,这些疾病是由于溶酶体蛋白基因的突变而发生的。戈谢病 (GD) 是一种常染色体隐性遗传病,也是最常见的 LSD 之一,是由于GBA1基因的双等位基因突变导致溶酶体酶酸-β-葡萄糖脑苷脂酶 (GCase) 缺乏引起的。降低的 GCase 活性导致葡萄糖神经酰胺 (GlcCer) 的积累,葡萄糖神经酰胺 (GlcCer) 被溶酶体酸性神经酰胺酶脱酰化为有毒代谢物葡萄糖基鞘氨醇 (GlcSph)。大多数GBA1变体在 ER 中被识别为错误折叠,其中重新折叠尝试的保留引发压力并激活称为未折叠蛋白质反应 (UPR) 的压力反应。
GD 的不同临床亚型基于是否主要累及中枢神经系统。1 型 GD (GD1) 是非神经病类型,然而,最近对 GD 与帕金森病发展相关性的认识违背了这一分类。GD1患者和GBA1携带者突变有发展为帕金森病表现的风险。帕金森病 (PD) 是第二大流行的神经退行性疾病,最终导致患者过早死亡的运动障碍。在 PD 和相关疾病(统称为突触核蛋白病)中,标志性病理学是称为路易体或路易神经突的 α-突触核蛋白阳性聚集体和多巴胺能神经元的死亡。虽然 PD 大多是散发的,但在约 5-10% 的病例中,这种疾病是由越来越多的基因中的致病变异引起的。PD最常见的遗传原因是GBA1突变。已经为这种联系提出了两种机制:(A)“功能获得”机制,其中突变的 GCase(蛋白质)有助于聚集体的形成和 PD 的发展,以及(B)“单倍体不足”(“失去功能”)模型,表明一个正常的 GBA1等位基因不足以携带足够的 GCase 活性,并且 GCase 的功能缺陷会阻碍 α-突触核蛋白代谢。溶酶体功能障碍、受损的自噬和线粒体自噬进一步增强了 α-突触核蛋白的积累,从而导致 PD 病理的发展。
本综述将详细阐述 GD 的生物学、其与 PD 和相关疾病的关联,并讨论这种关联的可能机制。










































 京公网安备 11010802027423号
京公网安备 11010802027423号