当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Binding Modes and Origins of Enantioselectivity in the Phase-Transfer-Catalyzed Conjugate Cyanation of β-Trifluoromethylated Chalcones
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-06-24 , DOI: 10.1021/acscatal.2c01904 Floris Buttard 1 , Pier Alexandre Champagne 1
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-06-24 , DOI: 10.1021/acscatal.2c01904 Floris Buttard 1 , Pier Alexandre Champagne 1
Affiliation
|
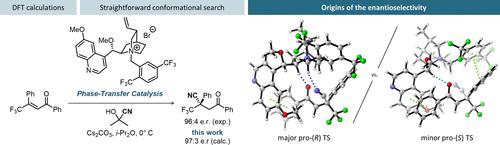
Despite their importance in industrial applications, enantioselective phase-transfer catalyzed reactions are poorly understood, especially for anionic nucleophiles other than enolates. In this context, we have studied the quinidinium-catalyzed phase-transfer cyanation of trifluoromethylated chalcones, reported by Shibata in 2012 and patented by Syngenta in 2016, using density functional theory (DFT) calculations. To efficiently sample the conformational space available to the noncovalently bound systems, we have designed a multistage procedure involving interrupted constrained optimizations, which identifies the most relevant conformers while sparing computational resources. A full study of the mechanism with a model catalyst confirmed that the 1,4-addition step is enantiodetermining in this reaction. Our calculations predict a 97:3 e.r. favoring the (R) enantiomer at the B3LYP-D3(BJ)/Def2TZVPP/SMD(i-Pr2O) level of theory, in excellent agreement with the experimental results. A complete analysis of the available binding modes of the substrates with the catalyst demonstrates that the strong stabilization of the two reaction partners by the ammonium α-hydrogens in the transition structure is critical to lower the activation barrier for the conjugate addition and that π-stacking interactions are not the main drivers of the selectivity as previously thought. These discoveries allowed us to propose a model of selectivity for the conjugate cyanation of chalcones, which mirrors the previously reported model for phase-transfer-catalyzed enolate alkylations.
中文翻译:

β-三氟甲基化查尔酮相转移催化共轭氰化中对映选择性的结合模式和来源
尽管它们在工业应用中很重要,但对映选择性相转移催化反应却知之甚少,尤其是对于烯醇化物以外的阴离子亲核试剂。在此背景下,我们使用密度泛函理论 (DFT) 计算研究了由 Shibata 于 2012 年报道并于 2016 年由先正达获得专利的三氟甲基化查尔酮的 quinidinium 催化相转移氰化。为了有效地对非共价结合系统可用的构象空间进行采样,我们设计了一个涉及中断约束优化的多阶段程序,该程序在节省计算资源的同时识别最相关的构象。使用模型催化剂对机理进行的全面研究证实,1,4-加成步骤在该反应中是对映体决定的。我们的计算预测为 97:3R ) 对映异构体在 B3LYP-D3(BJ)/Def2TZVPP/SMD( i -Pr 2 O) 理论水平,与实验结果非常吻合。对底物与催化剂的可用结合模式的完整分析表明,过渡结构中的 α-氢铵对两种反应伙伴的强稳定性对于降低共轭添加的活化势垒和 π 堆叠至关重要相互作用并不是以前认为的选择性的主要驱动力。这些发现使我们能够提出查尔酮共轭氰化选择性模型,这反映了先前报道的相转移催化烯醇烷基化模型。
更新日期:2022-06-24
中文翻译:
β-三氟甲基化查尔酮相转移催化共轭氰化中对映选择性的结合模式和来源
尽管它们在工业应用中很重要,但对映选择性相转移催化反应却知之甚少,尤其是对于烯醇化物以外的阴离子亲核试剂。在此背景下,我们使用密度泛函理论 (DFT) 计算研究了由 Shibata 于 2012 年报道并于 2016 年由先正达获得专利的三氟甲基化查尔酮的 quinidinium 催化相转移氰化。为了有效地对非共价结合系统可用的构象空间进行采样,我们设计了一个涉及中断约束优化的多阶段程序,该程序在节省计算资源的同时识别最相关的构象。使用模型催化剂对机理进行的全面研究证实,1,4-加成步骤在该反应中是对映体决定的。我们的计算预测为 97:3R ) 对映异构体在 B3LYP-D3(BJ)/Def2TZVPP/SMD( i -Pr 2 O) 理论水平,与实验结果非常吻合。对底物与催化剂的可用结合模式的完整分析表明,过渡结构中的 α-氢铵对两种反应伙伴的强稳定性对于降低共轭添加的活化势垒和 π 堆叠至关重要相互作用并不是以前认为的选择性的主要驱动力。这些发现使我们能够提出查尔酮共轭氰化选择性模型,这反映了先前报道的相转移催化烯醇烷基化模型。









































 京公网安备 11010802027423号
京公网安备 11010802027423号