当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mode selective chemistry for the dissociation of methane on efficient Ni/Pt-bimetallic alloy catalysts
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2022-06-24 , DOI: 10.1039/d2cp02030k Sudipta Roy 1 , Ashwani K Tiwari 1
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2022-06-24 , DOI: 10.1039/d2cp02030k Sudipta Roy 1 , Ashwani K Tiwari 1
Affiliation
|
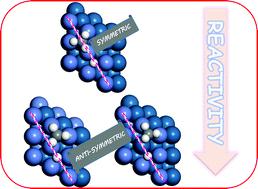
The mode selectivity of methane dissociation is studied on three different Ni/Pt-bimetallic alloy surfaces using a fully quantum approach based on reaction path Hamiltonian. Dissociative sticking probability depends on the composition of alloying metals, excited vibrational mode, and symmetry of the reaction path about the plane perpendicular to the catalyst surface containing the carbon atom and two hydrogen atoms. Our calculations show that symmetry of the minimum energy reaction path depends on the surface alloy composition. In the transition state, the dissociating C–H bond elongates significantly for the dissociation of methane on these alloy systems. A significant decrease in the frequency of the symmetric stretching mode and the two bending modes near the transition state is observed on all the alloy surfaces. Under the vibrational adiabatic limit, excitation of these softened modes enhanced the dissociation probability compared to the ground vibrational state. The reaction probability values decrease abruptly at the incident energies less than the zero-point energy corrected barrier height. With the inclusion of non-adiabatic vibrational coupling terms, reaction probability in the low incident energy region increases to a greater extent, and mode selective behavior also becomes different from that observed within the vibrational adiabatic limit. Symmetric stretching mode displayed the highest reactivity on all the alloy surfaces. Overall, Ni8/Pt(111) is found to be the most reactive toward the methane dissociation.
中文翻译:

高效 Ni/Pt 双金属合金催化剂上甲烷离解的模式选择化学
使用基于反应路径哈密顿量的全量子方法在三种不同的 Ni/Pt 双金属合金表面上研究了甲烷解离的模式选择性。解离粘附概率取决于合金金属的组成、激发的振动模式以及反应路径关于垂直于包含碳原子和两个氢原子的催化剂表面的平面的对称性。我们的计算表明,最小能量反应路径的对称性取决于表面合金成分。在过渡态,解离的 C-H 键显着拉长,以使甲烷在这些合金系统上解离。在所有合金表面上观察到对称拉伸模式和过渡态附近的两种弯曲模式的频率显着降低。在振动绝热极限下,与地面振动状态相比,这些软化模式的激发提高了解离概率。在小于零点能量校正势垒高度的入射能量处,反应概率值突然降低。随着非绝热振动耦合项的加入,低入射能区的反应概率更大程度地增加,并且模式选择行为也与在振动绝热极限内观察到的不同。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。在小于零点能量校正势垒高度的入射能量处,反应概率值突然降低。随着非绝热振动耦合项的加入,低入射能区的反应概率更大程度地增加,并且模式选择行为也与在振动绝热极限内观察到的不同。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。在小于零点能量校正势垒高度的入射能量处,反应概率值突然降低。随着非绝热振动耦合项的加入,低入射能区的反应概率更大程度地增加,并且模式选择行为也与在振动绝热极限内观察到的不同。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。并且模式选择行为也变得不同于在振动绝热极限内观察到的行为。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。并且模式选择行为也变得不同于在振动绝热极限内观察到的行为。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。
更新日期:2022-06-24
中文翻译:
高效 Ni/Pt 双金属合金催化剂上甲烷离解的模式选择化学
使用基于反应路径哈密顿量的全量子方法在三种不同的 Ni/Pt 双金属合金表面上研究了甲烷解离的模式选择性。解离粘附概率取决于合金金属的组成、激发的振动模式以及反应路径关于垂直于包含碳原子和两个氢原子的催化剂表面的平面的对称性。我们的计算表明,最小能量反应路径的对称性取决于表面合金成分。在过渡态,解离的 C-H 键显着拉长,以使甲烷在这些合金系统上解离。在所有合金表面上观察到对称拉伸模式和过渡态附近的两种弯曲模式的频率显着降低。在振动绝热极限下,与地面振动状态相比,这些软化模式的激发提高了解离概率。在小于零点能量校正势垒高度的入射能量处,反应概率值突然降低。随着非绝热振动耦合项的加入,低入射能区的反应概率更大程度地增加,并且模式选择行为也与在振动绝热极限内观察到的不同。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。在小于零点能量校正势垒高度的入射能量处,反应概率值突然降低。随着非绝热振动耦合项的加入,低入射能区的反应概率更大程度地增加,并且模式选择行为也与在振动绝热极限内观察到的不同。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。在小于零点能量校正势垒高度的入射能量处,反应概率值突然降低。随着非绝热振动耦合项的加入,低入射能区的反应概率更大程度地增加,并且模式选择行为也与在振动绝热极限内观察到的不同。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。并且模式选择行为也变得不同于在振动绝热极限内观察到的行为。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。并且模式选择行为也变得不同于在振动绝热极限内观察到的行为。对称拉伸模式在所有合金表面上显示出最高的反应性。总体而言,Ni8/Pt(111) 被发现对甲烷解离反应最为活跃。


























 京公网安备 11010802027423号
京公网安备 11010802027423号