Nature Machine Intelligence ( IF 18.8 ) Pub Date : 2022-06-23 , DOI: 10.1038/s42256-022-00501-8 Yuquan Li , Chang-Yu Hsieh , Ruiqiang Lu , Xiaoqing Gong , Xiaorui Wang , Pengyong Li , Shuo Liu , Yanan Tian , Dejun Jiang , Jiaxian Yan , Qifeng Bai , Huanxiang Liu , Shengyu Zhang , Xiaojun Yao
|
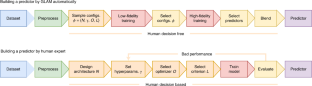
Improving drug discovery efficiency is a core and long-standing challenge in drug discovery. For this purpose, many graph learning methods have been developed to search potential drug candidates with fast speed and low cost. In fact, the pursuit of high prediction performance on a limited number of datasets has crystallized their architectures and hyperparameters, making them lose advantage in repurposing to new data generated in drug discovery. Here we propose a flexible method that can adapt to any dataset and make accurate predictions. The proposed method employs an adaptive pipeline to learn from a dataset and output a predictor. Without any manual intervention, the method achieves far better prediction performance on all tested datasets than traditional methods, which are based on hand-designed neural architectures and other fixed items. In addition, we found that the proposed method is more robust than traditional methods and can provide meaningful interpretability. Given the above, the proposed method can serve as a reliable method to predict molecular interactions and properties with high adaptability, performance, robustness and interpretability. This work takes a solid step forward to the purpose of aiding researchers to design better drugs with high efficiency.
中文翻译:
一种用于自动分子相互作用和性质预测的自适应图学习方法
提高药物发现效率是药物发现的核心和长期挑战。为此,已经开发了许多图学习方法来快速、低成本地搜索潜在的候选药物。事实上,在有限数量的数据集上追求高预测性能已经明确了它们的架构和超参数,使它们在重新利用药物发现中产生的新数据方面失去了优势。在这里,我们提出了一种灵活的方法,可以适应任何数据集并做出准确的预测。所提出的方法采用自适应管道从数据集中学习并输出预测器。在没有任何人工干预的情况下,该方法在所有测试数据集上实现了比基于手工设计的神经架构和其他固定项目的传统方法更好的预测性能。此外,我们发现所提出的方法比传统方法更稳健,并且可以提供有意义的可解释性。鉴于上述情况,所提出的方法可以作为一种可靠的方法来预测具有高适应性、性能、鲁棒性和可解释性的分子相互作用和性质。这项工作朝着帮助研究人员高效设计更好的药物的目的迈出了坚实的一步。









































 京公网安备 11010802027423号
京公网安备 11010802027423号