Biotechnology Advances ( IF 12.1 ) Pub Date : 2022-06-20 , DOI: 10.1016/j.biotechadv.2022.108009 Ondrej Vavra 1 , Jiri Damborsky 2 , David Bednar 1
|
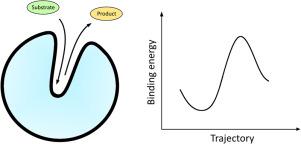
Acceleration of chemical reactions by the enzymes optimized using protein engineering represents one of the key pillars of the contribution of biotechnology towards sustainability. Tunnels and channels of enzymes with buried active sites enable the exchange of ligands, ions, and water molecules between the outer environment and active site pockets. The efficient exchange of ligands is a fundamental process of biocatalysis. Therefore, enzymes have evolved a wide range of mechanisms for repetitive conformational changes that enable periodic opening and closing. Protein-ligand interactions are traditionally studied by molecular docking, whereas molecular dynamics is the method of choice for studying conformational changes and ligand transport. However, computational demands make molecular dynamics impractical for screening purposes. Thus, several approximative methods have been recently developed to study interactions between a protein and ligand during the ligand transport process. Apart from identifying the best binding modes, these methods also provide information on the energetics of the transport and identify problematic regions limiting the ligand passage. These methods use approximations to simulate binding or unbinding events rapidly (calculation times from minutes to hours) and provide energy profiles that can be used to rank ligands or pathways. Here we provide a critical comparison of available methods, showcase their results on sample systems, discuss their practical applications in molecular biotechnologies and outline possible future developments.
中文翻译:
用于研究配体转运和合理设计用于生物技术的改进酶的快速近似方法
使用蛋白质工程优化的酶加速化学反应代表了生物技术对可持续性的贡献的关键支柱之一。具有埋藏活性位点的酶的隧道和通道能够在外部环境和活性位点口袋之间交换配体、离子和水分子。配体的有效交换是生物催化的基本过程。因此,酶已经进化出多种重复构象变化的机制,这些变化能够实现周期性的打开和关闭。传统上通过分子对接研究蛋白质-配体相互作用,而分子动力学是研究构象变化和配体转运的首选方法。然而,计算需求使得分子动力学对于筛选目的不切实际。因此,最近开发了几种近似方法来研究配体转运过程中蛋白质和配体之间的相互作用。除了确定最佳结合模式外,这些方法还提供有关运输能量学的信息,并确定限制配体通过的问题区域。这些方法使用近似值来快速模拟结合或解除结合事件(计算时间从几分钟到几小时),并提供可用于对配体或途径进行排序的能量分布。在这里,我们对可用方法进行了严格的比较,展示了它们在样品系统上的结果,讨论了它们在分子生物技术中的实际应用,并概述了未来可能的发展。










































 京公网安备 11010802027423号
京公网安备 11010802027423号