当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab initio trajectory surface-hopping dynamics studies of excited-state proton-coupled electron transfer reactions in trianisoleheptazine–phenol complexes
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-16 , DOI: 10.1039/d2cp01262f Xiang Huang 1 , Wolfgang Domcke 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-16 , DOI: 10.1039/d2cp01262f Xiang Huang 1 , Wolfgang Domcke 1
Affiliation
|
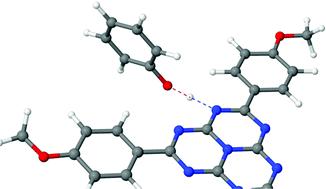
The excited-state proton-coupled electron-transfer (PCET) reaction in hydrogen-bonded complexes of trianisoleheptazine (TAHz), a chromophore related to polymeric carbon nitrides widely used in hydrogen-evolution photocatalysis, with several phenol derivatives were recently studied by Schlenker and coworkers with time-resolved photoluminescence quenching and pump–probe experiments. A pronounced dependence of the PCET reactivity on the electron-donating/electron-withdrawing character of the substituents on phenol was found, with indications of a barrierless or nearly barrierless PCET reaction for the most strongly electron-donating substituent, methoxy. In the present work, the excited-state PCET dynamics was explored with first-principles nonadiabatic dynamics simulations using the TDDFT/ωB97X-D electronic-structure model for two selected complexes, TAHz–phenol and TAHz–methoxyphenol. The qualitative reliability of the TDDFT/ωB97X-D electronic-structure model was assessed by extensive benchmarking of excitation energies and potential-energy profiles against a wave-function-based ab initio method, the algebraic-diagrammatic construction of second order (ADC(2)). The nonadiabatic dynamics simulations provide temporally and structurally resolved insights into paradigmatic PCET reactions in TAHz–phenol complexes. The radiationless relaxation of the photoexcited bright 1ππ* state to the long-lived dark S1 state of TAHz occurs in less than 100 fs. The ensuing PCET reaction on the adiabatic S1 surface is faster in TAHz–methoxyphenol complexes than in TAHz–phenol complexes due to a lower H-atom-transfer barrier, as observed in the experiments. The relaxation of the complexes to the electronic ground state is found to occur exclusively via PCET within the 250 fs time window covered by the present simulations, confirming the essential role of the hydrogen bond for the fluorescence quenching process. The absolute values of the computed PCET time constants are significantly shorter than those extracted from time-resolved photoluminescence measurements for mixtures of TAHz with phenolic substrates in toluene. The possible origins of this discrepancy are discussed.
中文翻译:

三苯甲醚庚嗪-苯酚配合物中激发态质子耦合电子转移反应的从头算轨迹表面跳跃动力学研究
Schlenker 和 Schlenker 最近研究了三苯甲异庚嗪 (TAHz) 的激发态质子耦合电子转移 (PCET) 反应,TAHz 是一种与广泛用于析氢光催化的聚合氮化碳相关的发色团,与几种苯酚衍生物与时间分辨光致发光猝灭和泵浦探针实验的同事。发现 PCET 反应性对苯酚上取代基的给电子/吸电子特性有显着依赖性,表明最强给电子取代基甲氧基的 PCET 反应是无屏障或几乎无屏障的。在目前的工作中,使用 TDDFT/ωB97X-D 电子结构模型对两个选定的配合物进行了第一性原理非绝热动力学模拟,探索了激发态 PCET 动力学,TAHz-苯酚和 TAHz-甲氧基苯酚。TDDFT/ωB97X-D 电子结构模型的定性可靠性通过对基于波函数的激发能量和势能分布进行广泛的基准测试来评估ab initio方法,二阶代数图解构造 (ADC(2))。非绝热动力学模拟为 TAHz-苯酚复合物中的典型 PCET 反应提供了时间和结构解析的见解。光激发的亮1 ππ* 态到 TAHz 的长寿命暗 S 1态的无辐射弛豫发生在小于 100 fs 的时间内。在绝热 S 1表面上随后发生的 PCET 反应在 TAHz-甲氧基苯酚复合物中比在 TAHz-苯酚复合物中更快,这是由于实验中观察到的较低的 H 原子转移势垒。发现配合物弛豫到电子基态仅通过以下方式发生本模拟涵盖的 250 fs 时间窗口内的 PCET,证实了氢键在荧光猝灭过程中的重要作用。计算出的 PCET 时间常数的绝对值明显短于从时间分辨光致发光测量中提取的 TAHz 与酚类底物在甲苯中的混合物的绝对值。讨论了这种差异的可能来源。
更新日期:2022-06-21
中文翻译:
三苯甲醚庚嗪-苯酚配合物中激发态质子耦合电子转移反应的从头算轨迹表面跳跃动力学研究
Schlenker 和 Schlenker 最近研究了三苯甲异庚嗪 (TAHz) 的激发态质子耦合电子转移 (PCET) 反应,TAHz 是一种与广泛用于析氢光催化的聚合氮化碳相关的发色团,与几种苯酚衍生物与时间分辨光致发光猝灭和泵浦探针实验的同事。发现 PCET 反应性对苯酚上取代基的给电子/吸电子特性有显着依赖性,表明最强给电子取代基甲氧基的 PCET 反应是无屏障或几乎无屏障的。在目前的工作中,使用 TDDFT/ωB97X-D 电子结构模型对两个选定的配合物进行了第一性原理非绝热动力学模拟,探索了激发态 PCET 动力学,TAHz-苯酚和 TAHz-甲氧基苯酚。TDDFT/ωB97X-D 电子结构模型的定性可靠性通过对基于波函数的激发能量和势能分布进行广泛的基准测试来评估ab initio方法,二阶代数图解构造 (ADC(2))。非绝热动力学模拟为 TAHz-苯酚复合物中的典型 PCET 反应提供了时间和结构解析的见解。光激发的亮1 ππ* 态到 TAHz 的长寿命暗 S 1态的无辐射弛豫发生在小于 100 fs 的时间内。在绝热 S 1表面上随后发生的 PCET 反应在 TAHz-甲氧基苯酚复合物中比在 TAHz-苯酚复合物中更快,这是由于实验中观察到的较低的 H 原子转移势垒。发现配合物弛豫到电子基态仅通过以下方式发生本模拟涵盖的 250 fs 时间窗口内的 PCET,证实了氢键在荧光猝灭过程中的重要作用。计算出的 PCET 时间常数的绝对值明显短于从时间分辨光致发光测量中提取的 TAHz 与酚类底物在甲苯中的混合物的绝对值。讨论了这种差异的可能来源。










































 京公网安备 11010802027423号
京公网安备 11010802027423号