当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Predicting Protein–Ligand Docking Structure with Graph Neural Network
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-06-14 , DOI: 10.1021/acs.jcim.2c00127 Huaipan Jiang 1 , Jian Wang 2 , Weilin Cong 1 , Yihe Huang 1 , Morteza Ramezani 1 , Anup Sarma 1 , Nikolay V Dokholyan 2, 3 , Mehrdad Mahdavi 1 , Mahmut T Kandemir 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-06-14 , DOI: 10.1021/acs.jcim.2c00127 Huaipan Jiang 1 , Jian Wang 2 , Weilin Cong 1 , Yihe Huang 1 , Morteza Ramezani 1 , Anup Sarma 1 , Nikolay V Dokholyan 2, 3 , Mehrdad Mahdavi 1 , Mahmut T Kandemir 1
Affiliation
|
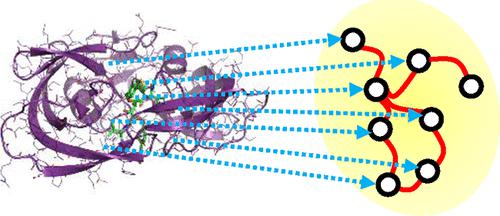
Modern day drug discovery is extremely expensive and time consuming. Although computational approaches help accelerate and decrease the cost of drug discovery, existing computational software packages for docking-based drug discovery suffer from both low accuracy and high latency. A few recent machine learning-based approaches have been proposed for virtual screening by improving the ability to evaluate protein–ligand binding affinity, but such methods rely heavily on conventional docking software to sample docking poses, which results in excessive execution latencies. Here, we propose and evaluate a novel graph neural network (GNN)-based framework, MedusaGraph, which includes both pose-prediction (sampling) and pose-selection (scoring) models. Unlike the previous machine learning-centric studies, MedusaGraph generates the docking poses directly and achieves from 10 to 100 times speedup compared to state-of-the-art approaches, while having a slightly better docking accuracy.
中文翻译:

用图神经网络预测蛋白质-配体对接结构
现代药物发现极其昂贵且耗时。尽管计算方法有助于加速和降低药物发现的成本,但用于基于对接的药物发现的现有计算软件包存在准确性低和延迟高的问题。最近提出了一些基于机器学习的方法,通过提高评估蛋白质-配体结合亲和力的能力来进行虚拟筛选,但这些方法严重依赖传统的对接软件来采样对接姿势,这导致执行延迟过大。在这里,我们提出并评估了一种基于图神经网络(GNN)的新型框架 MedusaGraph,其中包括姿势预测(采样)和姿势选择(评分)模型。与之前以机器学习为中心的研究不同,MedusaGraph 直接生成对接姿势,与最先进的方法相比,速度提高了 10 到 100 倍,同时具有稍好的对接精度。
更新日期:2022-06-14
中文翻译:
用图神经网络预测蛋白质-配体对接结构
现代药物发现极其昂贵且耗时。尽管计算方法有助于加速和降低药物发现的成本,但用于基于对接的药物发现的现有计算软件包存在准确性低和延迟高的问题。最近提出了一些基于机器学习的方法,通过提高评估蛋白质-配体结合亲和力的能力来进行虚拟筛选,但这些方法严重依赖传统的对接软件来采样对接姿势,这导致执行延迟过大。在这里,我们提出并评估了一种基于图神经网络(GNN)的新型框架 MedusaGraph,其中包括姿势预测(采样)和姿势选择(评分)模型。与之前以机器学习为中心的研究不同,MedusaGraph 直接生成对接姿势,与最先进的方法相比,速度提高了 10 到 100 倍,同时具有稍好的对接精度。








































 京公网安备 11010802027423号
京公网安备 11010802027423号