当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational mechanistic insights on Ag2O as a host for Li in lithium-ion batteries
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-13 , DOI: 10.1039/d2cp01674e C Hepsibah Priyadarshini 1 , V Sudha 1 , S Harinipriya 2
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-13 , DOI: 10.1039/d2cp01674e C Hepsibah Priyadarshini 1 , V Sudha 1 , S Harinipriya 2
Affiliation
|
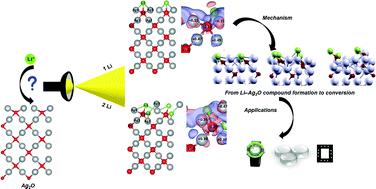
The lithiation mechanism of Ag2O with one and two Li atoms per unit cell carried out using density functional theory (DFT) studies indicate the adsorption of one Li atom at the tetrahedral interstitial site (TIS) as the most stable one with the formation of the Li–Ag2O compound. PDOS plots depict the hybridization of Li 2s with O 2p and Ag 5s states, resulting in the formation of both Li–O and Li–Ag bonds. Also, the Bader charge analysis validates the bonding of Li at TIS with both Ag and O atoms. The band structure plots showcase a surge in electronic conductivity, accounting for the metallic transition of Ag2O with the addition of Li. The difference in the charge distribution between the pairs of surface and subsurface silver atoms in charge density difference plots (CDDP) reveal the incorporation of Li, invoking charge inequivalence in addition to the symmetry inequivalence in the optimized structure. In the case of two Li atoms, Li placed on the top of oxygen (Otop) and at the subsurface of silver (Agss) was found to be the most stable structure with the formation of two strong Li–O bonds, indicating the feasibility of the conversion mechanism. The increase in the Bader charge of O (−1.28e) as well as CDDP establish the formation of Li2O by the conversion mechanism. Thus, the increase in the Li+ ion concentration becomes the deciding factor for the transition of the lithiation mechanism from Li–Ag2O compound formation to the conversion mechanism. The molecular dynamics (MD) simulations feature Li as an “antisite defect producing guest”, leading to the formation of Li–O bonds with a concomitant reduction in the number of Ag–O bonds as a function of time. This confirms a shift in the reaction kinetics from the formation of Li–Ag2O to the conversion mechanism.
中文翻译:

Ag2O 作为锂离子电池中锂的主体的计算机理见解
使用密度泛函理论 (DFT) 研究对每个晶胞具有一个和两个 Li 原子的 Ag 2 O 的锂化机制表明,一个 Li 原子在四面体间隙位 (TIS) 的吸附是最稳定的,并形成Li-Ag 2 O 化合物。PDOS 图描绘了 Li 2s 与 O 2p 和 Ag 5s 态的杂化,导致形成 Li-O 和 Li-Ag 键。此外,Bader 电荷分析验证了在 TIS 中 Li 与 Ag 和 O 原子的键合。能带结构图展示了电子电导率的激增,这说明了 Ag 2的金属跃迁O 加上 Li。电荷密度差图 (CDDP) 中表面和亚表面银原子对之间电荷分布的差异揭示了 Li 的结合,除了在优化结构中的对称不等价外,还引发了电荷不等价。在两个 Li 原子的情况下,Li 位于氧的顶部(O top)和银的亚表面(Ag ss)是最稳定的结构,形成了两个强的 Li-O 键,表明转换机制的可行性。O的Bader电荷(-1.28 e)以及CDDP的增加通过转化机制建立了Li 2 O的形成。因此,Li +离子浓度成为锂化机制从 Li-Ag 2 O 化合物形成转变为转化机制的决定因素。分子动力学(MD)模拟将Li作为“产生反位缺陷的客体”,导致Li-O键的形成,同时Ag-O键的数量随着时间的推移而减少。这证实了反应动力学从形成 Li-Ag 2 O 到转化机制的转变。
更新日期:2022-06-13
中文翻译:
Ag2O 作为锂离子电池中锂的主体的计算机理见解
使用密度泛函理论 (DFT) 研究对每个晶胞具有一个和两个 Li 原子的 Ag 2 O 的锂化机制表明,一个 Li 原子在四面体间隙位 (TIS) 的吸附是最稳定的,并形成Li-Ag 2 O 化合物。PDOS 图描绘了 Li 2s 与 O 2p 和 Ag 5s 态的杂化,导致形成 Li-O 和 Li-Ag 键。此外,Bader 电荷分析验证了在 TIS 中 Li 与 Ag 和 O 原子的键合。能带结构图展示了电子电导率的激增,这说明了 Ag 2的金属跃迁O 加上 Li。电荷密度差图 (CDDP) 中表面和亚表面银原子对之间电荷分布的差异揭示了 Li 的结合,除了在优化结构中的对称不等价外,还引发了电荷不等价。在两个 Li 原子的情况下,Li 位于氧的顶部(O top)和银的亚表面(Ag ss)是最稳定的结构,形成了两个强的 Li-O 键,表明转换机制的可行性。O的Bader电荷(-1.28 e)以及CDDP的增加通过转化机制建立了Li 2 O的形成。因此,Li +离子浓度成为锂化机制从 Li-Ag 2 O 化合物形成转变为转化机制的决定因素。分子动力学(MD)模拟将Li作为“产生反位缺陷的客体”,导致Li-O键的形成,同时Ag-O键的数量随着时间的推移而减少。这证实了反应动力学从形成 Li-Ag 2 O 到转化机制的转变。










































 京公网安备 11010802027423号
京公网安备 11010802027423号