当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A caveat of the charge-extrapolation scheme for modeling electrochemical reactions on semiconductor surfaces: an issue induced by a discontinuous Fermi level change
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-10 , DOI: 10.1039/d2cp00642a Yu Liu 1 , Xinlong Ding 2 , Mohan Chen 1 , Shenzhen Xu 3
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-10 , DOI: 10.1039/d2cp00642a Yu Liu 1 , Xinlong Ding 2 , Mohan Chen 1 , Shenzhen Xu 3
Affiliation
|
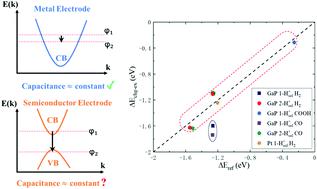
(Photo)electrochemical surface reactions in realistic experimental systems occur under a constant-potential condition, while the ab initio simulations of electrochemical reactions are mostly performed under a constant-charge condition. A charge-extrapolation scheme proposed by earlier theoretical studies converts constant-charge reaction energies to constant-potential reaction energies for electrochemical reactions on metal surfaces, which is based on a capacitor-model assumption to approximate the surface electrical double layer. However, the charge-extrapolation approach may be problematic when applied to models of photoelectrochemical reactions on semiconductor surfaces with a cross-bandgap Fermi level change along the reaction path. We perform density-functional-theory calculations to show that the error is induced by an abrupt change of the modeling system's potential making the capacitor model assumption invalid. We further propose an approach to avoid the cross-bandgap Fermi level change in the simulations of semiconductor surface reactions, with which the charge-extrapolation scheme still can be employed to compute the constant-potential reaction energies for the semiconductor photoelectrode cases.
中文翻译:

用于模拟半导体表面电化学反应的电荷外推方案的警告:由不连续费米能级变化引起的问题
现实实验系统中的(光)电化学表面反应发生在恒电位条件下,而从头算电化学反应的模拟大多在恒定电荷条件下进行。早期理论研究提出的电荷外推方案将恒定电荷反应能转换为金属表面电化学反应的恒定电位反应能,该方案基于电容器模型假设来近似表面双电层。然而,当应用于半导体表面上的光电化学反应模型时,电荷外推方法可能会出现问题,并且跨带隙费米能级沿反应路径发生变化。我们进行了密度泛函理论计算,以表明该误差是由建模系统电位的突然变化引起的,从而使电容器模型假设无效。
更新日期:2022-06-10
中文翻译:
用于模拟半导体表面电化学反应的电荷外推方案的警告:由不连续费米能级变化引起的问题
现实实验系统中的(光)电化学表面反应发生在恒电位条件下,而从头算电化学反应的模拟大多在恒定电荷条件下进行。早期理论研究提出的电荷外推方案将恒定电荷反应能转换为金属表面电化学反应的恒定电位反应能,该方案基于电容器模型假设来近似表面双电层。然而,当应用于半导体表面上的光电化学反应模型时,电荷外推方法可能会出现问题,并且跨带隙费米能级沿反应路径发生变化。我们进行了密度泛函理论计算,以表明该误差是由建模系统电位的突然变化引起的,从而使电容器模型假设无效。








































 京公网安备 11010802027423号
京公网安备 11010802027423号