Frontiers of Chemical Science and Engineering ( IF 4.5 ) Pub Date : 2022-06-10 , DOI: 10.1007/s11705-022-2169-8 Juntian Niu , Haiyu Liu , Yan Jin , Baoguo Fan , Wenjie Qi , Jingyu Ran
|
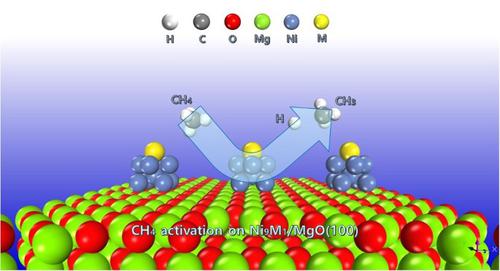
Methane activation is a pivotal step in the application of natural gas converting into high-value added chemicals via methane steam/dry reforming reactions. Ni element was found to be the most widely used catalyst. In present work, methane activation on MgO supported Ni-M (M = Fe, Co, Cu, Pd, Pt) cluster was explored through detailed density functional theory calculations, compared to pure Ni cluster. CH4 adsorption on Cu promoted Ni cluster requires overcoming an energy of 0.07 eV, indicating that it is slightly endothermic and unfavored to occur, while the adsorption energies of other promoters M (M = Fe, Co, Pd and Pt) are all higher than that of pure Ni cluster. The role of M on the first C-H bond cleavage of CH4 was investigated. Doping elements of the same period in Ni cluster, such as Fe, Co and Cu, for C-H bond activation follows the trend of the decrease of metal atom radius. As a result, Ni-Fe shows the best ability for C-H bond cleavage. In addition, doping the elements of the same family, like Pd and Pt, for CH4 activation is according to the increase of metal atom radius. Consequently, C-H bond activation demands a lower energy barrier on Ni-Pt cluster. To illustrate the adsorptive dissociation behaviors of CH4 at different Ni-M clusters, the Mulliken atomic charge was analyzed. In general, the electron gain of CH4 binding at different Ni-M clusters follows the sequence of Ni-Cu (−0.02 e) < Ni (−0.04 e) < Ni-Pd (−0.08 e) < Ni-Pt (−0.09 e) < Ni-Co (−0.10 e) < Ni-Fe (−0.12 e), and the binding strength between catalysts and CH4 raises with the CH4 electron gain increasing. This work provides insights into understanding the role of promoter metal M on thermal-catalytic activation of CH4 over Ni/MgO catalysts, and is useful to interpret the reaction at an atomic scale.
中文翻译:
MgO负载Ni9M1簇上甲烷活化的密度泛函理论研究:M对CH活化的作用
甲烷活化是应用天然气通过甲烷蒸汽/干重整反应转化为高附加值化学品的关键步骤。Ni元素被发现是最广泛使用的催化剂。在目前的工作中,与纯 Ni 簇相比,通过详细的密度泛函理论计算探索了 MgO 负载的 Ni-M(M = Fe、Co、Cu、Pd、Pt)簇上的甲烷活化。Cu促进的Ni簇对CH 4的吸附需要克服0.07 eV的能量,表明它是轻微吸热的,不利于发生,而其他促进剂M(M = Fe、Co、Pd和Pt)的吸附能均高于纯镍簇。M在CH 4的第一个CH键断裂中的作用进行了调查。在Ni团簇中掺杂Fe、Co、Cu等同期元素对CH键活化的作用遵循金属原子半径减小的趋势。结果,Ni-Fe显示出最好的CH键断裂能力。此外,掺杂同族元素,如Pd和Pt,激活CH 4是根据金属原子半径的增加。因此,CH 键活化需要 Ni-Pt 簇上的较低能垒。为了说明 CH 4在不同 Ni-M 簇上的吸附解离行为,分析了 Mulliken 原子电荷。一般来说,CH 4的电子增益在不同 Ni-M 簇上的结合遵循 Ni-Cu (-0.02 e) < Ni (-0.04 e) < Ni-Pd (-0.08 e) < Ni-Pt (-0.09 e) < Ni-Co (- 0.10 e) < Ni-Fe (-0.12 e),催化剂与CH 4的结合强度随着CH 4电子增益的增加而提高。这项工作为理解助催化剂金属 M 在 Ni/MgO 催化剂上对 CH 4的热催化活化的作用提供了见解,并有助于在原子尺度上解释反应。


























 京公网安备 11010802027423号
京公网安备 11010802027423号