当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Liquid-like properties of cyclopentadienyl complexes of barium: molecular dynamics simulations of nanoscale droplets
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-09 , DOI: 10.1039/d2cp02322a Hye Ree Hyun 1 , Jungim Han 2 , Juhyung Lim 2 , Young Jae Park 2 , Byoungki Choi 2 , Chul Baik 2 , Jun Soo Kim 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-06-09 , DOI: 10.1039/d2cp02322a Hye Ree Hyun 1 , Jungim Han 2 , Juhyung Lim 2 , Young Jae Park 2 , Byoungki Choi 2 , Chul Baik 2 , Jun Soo Kim 1
Affiliation
|
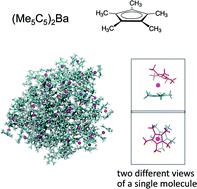
Cyclopentadienyl complexes of barium have great utility in materials science and engineering, in particular, as precursors in the atomic layer deposition processes, which are required to be fluidic as well as thermally stable and volatile. Here, we investigated the liquid-like properties of cyclopentadienyl barium complexes including (Me5C5)2Ba, (tBu3C5H2)2Ba, (iPr4C5H)2Ba, (iPr5C5)2Ba, and [(SiMe3)3C5H2]2Ba, using molecular dynamics simulations of nanoscale droplets. The compounds were modeled using a recently developed generic force field, GFN-FF. Nanoscale droplets with about 5.0 nm diameters were formed by aggregating 96 molecules of each compound. Simulation results reveal that substituting methyl groups of (Me5C5)2Ba with other alkyl and silyl moieties has a non-negligible effect on the intra- and intermolecular structure and dynamics. In particular, in contrast to more flexible (Me5C5)2Ba, the substitution with five iso-propyl groups to form (iPr5C5)2Ba adds rigidity to the complex with restricted orientational fluctuations for two cyclopentadienyl ligands and arranges molecules parallel to each other with greater probability. In addition, comparison between (tBu3C5H2)2Ba, with three tert-butyl groups, and its silyl analogue, [(SiMe3)3C5H2]2Ba, reveals that intermolecular interactions between the molecules with silyl groups are softer than those with tert-butyl groups and result in broader radial distribution functions, whereas the dynamic properties are similar for both compounds. This work suggests that molecular dynamics simulations contribute to molecular-level understanding of the effect of chemical substitution in organometallic compounds on the intra- and intermolecular properties of molecular liquids.
中文翻译:

钡的环戊二烯基配合物的类液体特性:纳米级液滴的分子动力学模拟
钡的环戊二烯基配合物在材料科学和工程中具有很大的用途,特别是作为原子层沉积过程中的前体,要求其具有流动性以及热稳定性和挥发性。在这里,我们研究了包括 (Me 5 C 5 ) 2 Ba, ( t Bu 3 C 5 H 2 ) 2 Ba, ( i Pr 4 C 5 H) 2 Ba, ( i Pr 5 )的环戊二烯基钡配合物的类液体性质。C 5 ) 2 Ba 和 [(SiMe 3 )3 C 5 H 2 ] 2 Ba,使用纳米级液滴的分子动力学模拟。使用最近开发的通用力场 GFN-FF 对化合物进行建模。通过聚集每种化合物的 96 个分子形成直径约为 5.0 nm 的纳米级液滴。模拟结果表明,用其他烷基和甲硅烷基部分取代(Me 5 C 5 ) 2 Ba 的甲基对分子内和分子间结构和动力学具有不可忽略的影响。特别地,与更灵活的 (Me 5 C 5 ) 2 Ba 相比,用五个异丙基取代形成 ( i Pr5 C 5 ) 2 Ba 为配合物增加了刚性,两个环戊二烯基配体的取向波动受限,并使分子彼此平行排列的可能性更大。此外,具有三个叔丁基的 ( t Bu 3 C 5 H 2 ) 2 Ba与其甲硅烷基类似物 [(SiMe 3 ) 3 C 5 H 2 ] 2 Ba 之间的比较揭示了分子之间的分子间相互作用具有甲硅烷基的比具有叔丁基的更柔软-丁基基团并导致更广泛的径向分布函数,而两种化合物的动态特性相似。这项工作表明,分子动力学模拟有助于在分子水平上理解有机金属化合物中的化学取代对分子液体的分子内和分子间性质的影响。
更新日期:2022-06-09
中文翻译:
钡的环戊二烯基配合物的类液体特性:纳米级液滴的分子动力学模拟
钡的环戊二烯基配合物在材料科学和工程中具有很大的用途,特别是作为原子层沉积过程中的前体,要求其具有流动性以及热稳定性和挥发性。在这里,我们研究了包括 (Me 5 C 5 ) 2 Ba, ( t Bu 3 C 5 H 2 ) 2 Ba, ( i Pr 4 C 5 H) 2 Ba, ( i Pr 5 )的环戊二烯基钡配合物的类液体性质。C 5 ) 2 Ba 和 [(SiMe 3 )3 C 5 H 2 ] 2 Ba,使用纳米级液滴的分子动力学模拟。使用最近开发的通用力场 GFN-FF 对化合物进行建模。通过聚集每种化合物的 96 个分子形成直径约为 5.0 nm 的纳米级液滴。模拟结果表明,用其他烷基和甲硅烷基部分取代(Me 5 C 5 ) 2 Ba 的甲基对分子内和分子间结构和动力学具有不可忽略的影响。特别地,与更灵活的 (Me 5 C 5 ) 2 Ba 相比,用五个异丙基取代形成 ( i Pr5 C 5 ) 2 Ba 为配合物增加了刚性,两个环戊二烯基配体的取向波动受限,并使分子彼此平行排列的可能性更大。此外,具有三个叔丁基的 ( t Bu 3 C 5 H 2 ) 2 Ba与其甲硅烷基类似物 [(SiMe 3 ) 3 C 5 H 2 ] 2 Ba 之间的比较揭示了分子之间的分子间相互作用具有甲硅烷基的比具有叔丁基的更柔软-丁基基团并导致更广泛的径向分布函数,而两种化合物的动态特性相似。这项工作表明,分子动力学模拟有助于在分子水平上理解有机金属化合物中的化学取代对分子液体的分子内和分子间性质的影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号