当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Assessment of density functional approximations for N2 and CO2 physisorption on benzene and graphene
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-06-06 , DOI: 10.1002/jcc.26945 Víctor M Rayón 1 , Iván Cabria 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-06-06 , DOI: 10.1002/jcc.26945 Víctor M Rayón 1 , Iván Cabria 2
Affiliation
|
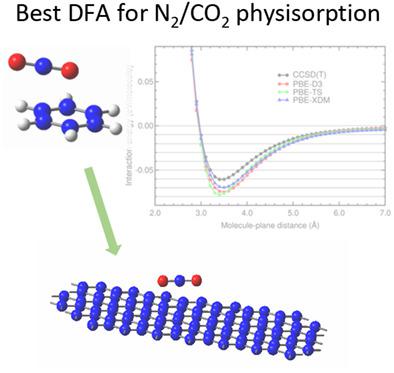
Experimental isotherms of N2 and CO2 on carbon-based porous materials and models of the physisorption of gases on surfaces are used to obtain the pore size distribution (PSD). An accurate modelization of the physisorption of N2 and CO2 on the surface of carbon-based porous materials is important to obtain accurate N2 and CO2 storage capacities and reliable PSDs. Physisorption depends on the dispersion interactions. High precision ab initio methods, such as CCSD(T), consider accurately the dispersion interactions, but they are computationally expensive. Double hybrid, hybrid and DFT-based methods are much less expensive. In the case of graphene, there are experimental data of the adsorption of N2 and CO2 on graphite that can be used to build the Steele interaction potential of these gases on graphene. The goal is to find out hybrid and/or DFT methods that are as accurate as the CCSD(T) on benzene and as accurate as the experimental results on graphene. Calculations of the interaction energy curves of N2 and CO2 on benzene and graphene have been carried out using the CCSD(T) method and several double hybrid, hybrid, and DFT methods that consider the dispersion interactions. The energy curves on benzene have been compared to the CCSD(T) and the energy curves on graphene have been compared with the Steele energy curves. The comparisons indicate that double hybrids with dispersion corrections and ωB97 based DFT methods are accurate enough for benzene. For graphene, only the PBE-XDM functional has a good agreement with the Steele energy curves.
中文翻译:

苯和石墨烯上 N2 和 CO2 物理吸附的密度泛函近似评估
使用碳基多孔材料上的N 2和CO 2实验等温线以及表面上气体的物理吸附模型来获得孔径分布(PSD)。碳基多孔材料表面上N 2和CO 2物理吸附的准确建模对于获得准确的N 2和CO 2存储容量和可靠的PSD非常重要。物理吸附取决于分散相互作用。 CCSD(T) 等高精度从头计算方法可以准确地考虑色散相互作用,但计算成本较高。双混合、混合和基于 DFT 的方法要便宜得多。就石墨烯而言,石墨上吸附N 2和CO 2的实验数据可用于构建这些气体在石墨烯上的斯蒂尔相互作用势。目标是找到与苯上的 CCSD(T) 一样准确以及与石墨烯上的实验结果一样准确的混合和/或 DFT 方法。使用CCSD(T)方法和几种考虑色散相互作用的双杂化、杂化和DFT方法计算了N 2和CO 2在苯和石墨烯上的相互作用能曲线。苯的能量曲线与 CCSD(T) 进行了比较,石墨烯的能量曲线与 Steele 能量曲线进行了比较。比较表明,具有色散校正的双杂化和基于ω B97 的 DFT 方法对于苯来说足够准确。对于石墨烯,只有 PBE-XDM 泛函与 Steele 能量曲线具有良好的一致性。
更新日期:2022-06-06
中文翻译:
苯和石墨烯上 N2 和 CO2 物理吸附的密度泛函近似评估
使用碳基多孔材料上的N 2和CO 2实验等温线以及表面上气体的物理吸附模型来获得孔径分布(PSD)。碳基多孔材料表面上N 2和CO 2物理吸附的准确建模对于获得准确的N 2和CO 2存储容量和可靠的PSD非常重要。物理吸附取决于分散相互作用。 CCSD(T) 等高精度从头计算方法可以准确地考虑色散相互作用,但计算成本较高。双混合、混合和基于 DFT 的方法要便宜得多。就石墨烯而言,石墨上吸附N 2和CO 2的实验数据可用于构建这些气体在石墨烯上的斯蒂尔相互作用势。目标是找到与苯上的 CCSD(T) 一样准确以及与石墨烯上的实验结果一样准确的混合和/或 DFT 方法。使用CCSD(T)方法和几种考虑色散相互作用的双杂化、杂化和DFT方法计算了N 2和CO 2在苯和石墨烯上的相互作用能曲线。苯的能量曲线与 CCSD(T) 进行了比较,石墨烯的能量曲线与 Steele 能量曲线进行了比较。比较表明,具有色散校正的双杂化和基于ω B97 的 DFT 方法对于苯来说足够准确。对于石墨烯,只有 PBE-XDM 泛函与 Steele 能量曲线具有良好的一致性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号