Analytica Chimica Acta ( IF 5.7 ) Pub Date : 2022-06-07 , DOI: 10.1016/j.aca.2022.340037 Yanting Guo 1 , Dahang Yu 1 , Kellye A Cupp-Sutton 1 , Xiaowen Liu 2 , Si Wu 1
|
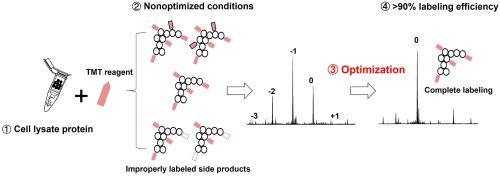
Isobaric chemical tag labels (e.g., iTRAQ and TMT) have been extensively utilized as a standard quantification approach in bottom-up proteomics, which provides high multiplexing capacity and enables MS2-level quantification while not complicating the MS1 scans. We recently demonstrated the feasibility of intact protein TMT labeling for the identification and quantification with top-down proteomics of smaller intact proteoforms (<35 kDa) in complex biological samples through the removal of large proteins prior to labeling. Still, the production of side products during TMT labeling (i.e., incomplete labeling or labeling of unintended residues) complicated the analysis of complex protein samples. In this study, we systematically evaluated the protein-level TMT labeling reaction parameters, including TMT-to-protein mass ratio, pH/concentration of quenching buffer, protein concentration, reaction time, and reaction buffer. Our results indicated that: (1) high TMT-to-protein mass ratio (e.g., 8:1, 4:1), (2) high pH/concentration of quenching buffer (pH > 9.1, final hydroxylamine concentration >0.3%), and (3) high protein concentration (e.g., > 1.0 μg/μL) resulted in optimal labeling efficiency and minimized production of over/underlabeled side products. >90% labeling efficiency was achieved for E. coli cell lysate after optimization of protein-level TMT labeling conditions. In addition, a double labeling approach was developed for efficiently labeling limited biological samples with low concentrations. This research provides practical guidance for efficient TMT labeling of complex intact protein samples, which can be readily adopted in the high-throughput quantification top-down proteomics.
中文翻译:
通过自上而下的蛋白质组学优化复杂样品中蛋白质水平串联质量标签 (TMT) 标记条件
同量化学标签(例如iTRAQ 和 TMT)已广泛用作自下而上蛋白质组学中的标准定量方法,其提供高多重能力并实现 MS2 级定量,同时不会使 MS1 扫描复杂化。我们最近证明了完整蛋白质 TMT 标记的可行性,通过在标记前去除大蛋白质,通过自上而下的蛋白质组学对复杂生物样品中较小的完整蛋白质形式 (<35 kDa) 进行识别和定量。尽管如此,TMT 标记过程中副产物的产生(即标记不完整或标记非预期残留物)使复杂蛋白质样品的分析变得复杂。在本研究中,我们系统地评估了蛋白质水平的TMT标记反应参数,包括TMT与蛋白质的质量比、淬灭缓冲液的pH/浓度、蛋白质浓度、反应时间和反应缓冲液。我们的结果表明:(1)高TMT与蛋白质质量比(例如8:1、4:1),(2)高pH/淬灭缓冲液浓度(pH > 9.1,最终羟胺浓度> 0.3%) ,(3) 高蛋白质浓度(例如, > 1.0 μg/μL)可实现最佳标记效率并最大程度地减少标记过度/标记不足的副产物的产生。优化蛋白质水平 TMT 标记条件后,大肠杆菌细胞裂解物的标记效率达到了 >90% 。此外,还开发了一种双标记方法,用于有效标记低浓度的有限生物样品。这项研究为复杂完整蛋白质样品的高效TMT标记提供了实用指导,可以很容易地应用于高通量定量自上而下的蛋白质组学中。










































 京公网安备 11010802027423号
京公网安备 11010802027423号