当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Spin–Orbit Couplings for Nonadiabatic Molecular Dynamics at the ΔSCF Level
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-06-06 , DOI: 10.1021/acs.jctc.1c01046 Momir Mališ 1 , Eva Vandaele 1 , Sandra Luber 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-06-06 , DOI: 10.1021/acs.jctc.1c01046 Momir Mališ 1 , Eva Vandaele 1 , Sandra Luber 1
Affiliation
|
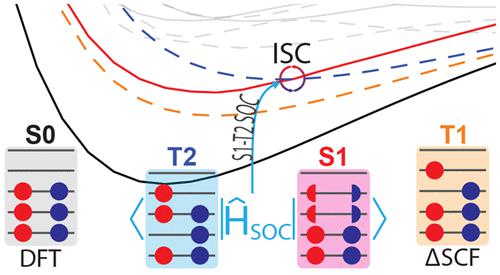
A procedure for the calculation of spin–orbit coupling (SOC) at the delta self-consistent field (ΔSCF) level of theory is presented. Singlet and triplet excited electronic states obtained with the ΔSCF method are expanded into a linear combination of singly excited Slater determinants composed of ground electronic state Kohn–Sham orbitals. This alleviates the nonorthogonality between excited and ground electronic states and introduces a framework, similar to the auxiliary wave function at the time-dependent density functional theory (TD-DFT) level, for the calculation of observables. The ΔSCF observables of the formaldehyde system were compared to reference TD-DFT values. Our procedure gives all components (energies, gradients, nonadiabatic couplings, and SOC terms) at the ΔSCF level of theory for conducting efficient, full-atomistic nonadiabatic molecular dynamics with intersystem crossing, particularly in condensed phase systems.
中文翻译:

ΔSCF 水平的非绝热分子动力学的自旋轨道耦合
提出了一种在增量自洽场 (ΔSCF) 理论水平上计算自旋轨道耦合 (SOC) 的程序。用 ΔSCF 方法获得的单重和三重激发电子态被扩展为由基态电子态 Kohn-Sham 轨道组成的单激发 Slater 行列式的线性组合。这减轻了激发态和基态电子态之间的非正交性,并引入了一个类似于时间相关密度泛函理论 (TD-DFT) 级别的辅助波函数的框架,用于计算可观察量。将甲醛系统的 ΔSCF 观测值与参考 TD-DFT 值进行比较。我们的程序在 ΔSCF 理论水平上给出了所有分量(能量、梯度、非绝热耦合和 SOC 项),以实现高效、
更新日期:2022-06-06
中文翻译:
ΔSCF 水平的非绝热分子动力学的自旋轨道耦合
提出了一种在增量自洽场 (ΔSCF) 理论水平上计算自旋轨道耦合 (SOC) 的程序。用 ΔSCF 方法获得的单重和三重激发电子态被扩展为由基态电子态 Kohn-Sham 轨道组成的单激发 Slater 行列式的线性组合。这减轻了激发态和基态电子态之间的非正交性,并引入了一个类似于时间相关密度泛函理论 (TD-DFT) 级别的辅助波函数的框架,用于计算可观察量。将甲醛系统的 ΔSCF 观测值与参考 TD-DFT 值进行比较。我们的程序在 ΔSCF 理论水平上给出了所有分量(能量、梯度、非绝热耦合和 SOC 项),以实现高效、








































 京公网安备 11010802027423号
京公网安备 11010802027423号