当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Combined Deep Learning and Classical Potential Approach for Modeling Diffusion in UiO-66
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-06-02 , DOI: 10.1021/acs.jctc.2c00010 Siddarth K Achar 1 , Jacob J Wardzala 2 , Leonardo Bernasconi 3 , Linfeng Zhang 4, 5 , J Karl Johnson 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-06-02 , DOI: 10.1021/acs.jctc.2c00010 Siddarth K Achar 1 , Jacob J Wardzala 2 , Leonardo Bernasconi 3 , Linfeng Zhang 4, 5 , J Karl Johnson 2
Affiliation
|
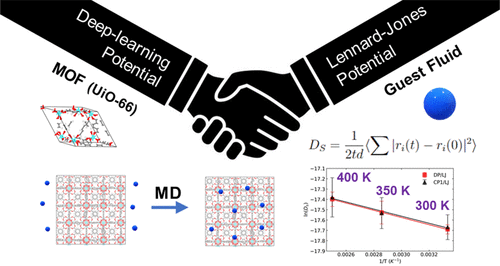
Modeling of diffusion of adsorbates through porous materials with atomistic molecular dynamics (MD) can be a challenging task if the flexibility of the adsorbent needs to be included. This is because potentials need to be developed that accurately account for the motion of the adsorbent in response to the presence of adsorbate molecules. In this work, we show that it is possible to use accurate machine learning atomistic potentials for metal–organic frameworks in concert with classical potentials for adsorbates to accurately compute diffusivities though a hybrid potential approach. As a proof-of-concept, we have developed an accurate deep learning potential (DP) for UiO-66, a metal–organic framework, and used this DP to perform hybrid potential simulations, modeling diffusion of neon and xenon through the crystal. The adsorbate–adsorbate interactions were modeled with Lennard–Jones (LJ) potentials, the adsorbent–adsorbent interactions were described by the DP, and the adsorbent–adsorbate interactions used LJ cross-interactions. Thus, our hybrid potential allows for adsorbent–adsorbate interactions with classical potentials but models the response of the adsorbent to the presence of the adsorbate through near-DFT accuracy DPs. This hybrid approach does not require refitting the DP for new adsorbates. We calculated self-diffusion coefficients for Ne in UiO-66 from DFT-MD, our hybrid DP/LJ approach, and from two different classical potentials for UiO-66. Our DP/LJ results are in excellent agreement with DFT-MD. We modeled diffusion of Xe in UiO-66 with DP/LJ and a classical potential. Diffusion of Xe in UiO-66 is about a factor of 30 slower than that of Ne, so it is not computationally feasible to compute Xe diffusion with DFT-MD. Our hybrid DP–classical potential approach can be applied to other MOFs and other adsorbates, making it possible to use an accurate DP generated from DFT simulations of an empty adsorbent in concert with existing classical potentials for adsorbates to model adsorption and diffusion within the porous material, including adsorbate-induced changes to the framework.
中文翻译:

UiO-66 中用于建模扩散的深度学习和经典势能方法相结合
如果需要包括吸附剂的灵活性,则使用原子分子动力学 (MD) 模拟吸附物通过多孔材料的扩散可能是一项具有挑战性的任务。这是因为需要开发能够准确解释吸附剂响应于吸附质分子的存在而运动的电位。在这项工作中,我们表明可以使用金属有机框架的准确机器学习原子势与吸附物的经典势相一致,通过混合势方法准确计算扩散率。作为概念验证,我们为金属有机框架 UiO-66 开发了准确的深度学习电位 (DP),并使用该 DP 进行混合电位模拟,模拟氖和氙通过晶体的扩散。吸附质-吸附质相互作用用 Lennard-Jones (LJ) 电位建模,吸附剂-吸附剂相互作用由 DP 描述,吸附剂-吸附质相互作用使用 LJ 交叉相互作用。因此,我们的混合势允许吸附剂-吸附质与经典势能的相互作用,但通过近 DFT 精度 DP 模拟吸附剂对吸附质存在的响应。这种混合方法不需要为新的吸附物改装 DP。我们从 DFT-MD、我们的混合 DP/LJ 方法以及 UiO-66 的两个不同经典势能计算了 UiO-66 中 Ne 的自扩散系数。我们的 DP/LJ 结果与 DFT-MD 非常一致。我们用 DP/LJ 和经典势模拟了 Xe 在 UiO-66 中的扩散。UiO-66 中 Xe 的扩散比 Ne 慢 30 倍,因此用 DFT-MD 计算 Xe 扩散在计算上是不可行的。我们的混合 DP-经典势能方法可应用于其他 MOF 和其他吸附物,从而可以使用由空吸附剂的 DFT 模拟产生的准确 DP 与现有的吸附物经典势相一致来模拟多孔材料内的吸附和扩散,包括吸附质引起的框架变化。
更新日期:2022-06-02
中文翻译:
UiO-66 中用于建模扩散的深度学习和经典势能方法相结合
如果需要包括吸附剂的灵活性,则使用原子分子动力学 (MD) 模拟吸附物通过多孔材料的扩散可能是一项具有挑战性的任务。这是因为需要开发能够准确解释吸附剂响应于吸附质分子的存在而运动的电位。在这项工作中,我们表明可以使用金属有机框架的准确机器学习原子势与吸附物的经典势相一致,通过混合势方法准确计算扩散率。作为概念验证,我们为金属有机框架 UiO-66 开发了准确的深度学习电位 (DP),并使用该 DP 进行混合电位模拟,模拟氖和氙通过晶体的扩散。吸附质-吸附质相互作用用 Lennard-Jones (LJ) 电位建模,吸附剂-吸附剂相互作用由 DP 描述,吸附剂-吸附质相互作用使用 LJ 交叉相互作用。因此,我们的混合势允许吸附剂-吸附质与经典势能的相互作用,但通过近 DFT 精度 DP 模拟吸附剂对吸附质存在的响应。这种混合方法不需要为新的吸附物改装 DP。我们从 DFT-MD、我们的混合 DP/LJ 方法以及 UiO-66 的两个不同经典势能计算了 UiO-66 中 Ne 的自扩散系数。我们的 DP/LJ 结果与 DFT-MD 非常一致。我们用 DP/LJ 和经典势模拟了 Xe 在 UiO-66 中的扩散。UiO-66 中 Xe 的扩散比 Ne 慢 30 倍,因此用 DFT-MD 计算 Xe 扩散在计算上是不可行的。我们的混合 DP-经典势能方法可应用于其他 MOF 和其他吸附物,从而可以使用由空吸附剂的 DFT 模拟产生的准确 DP 与现有的吸附物经典势相一致来模拟多孔材料内的吸附和扩散,包括吸附质引起的框架变化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号