当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Evaluating Geometric Definitions of Stacking for RNA Dinucleoside Monophosphates Using Molecular Mechanics Calculations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-06-02 , DOI: 10.1021/acs.jctc.2c00178 Amirhossein Taghavi 1, 2 , Ivan Riveros 1 , David J Wales 3 , Ilyas Yildirim 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-06-02 , DOI: 10.1021/acs.jctc.2c00178 Amirhossein Taghavi 1, 2 , Ivan Riveros 1 , David J Wales 3 , Ilyas Yildirim 1
Affiliation
|
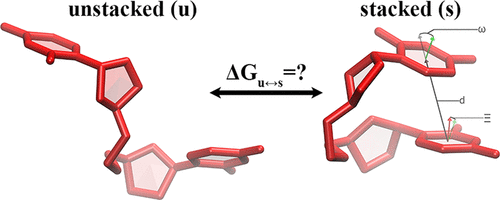
RNA modulation via small molecules is a novel approach in pharmacotherapies, where the determination of the structural properties of RNA motifs is considered a promising way to develop drugs capable of targeting RNA structures to control diseases. However, due to the complexity and dynamic nature of RNA molecules, the determination of RNA structures using experimental approaches is not always feasible, and computational models employing force fields can provide important insight. The quality of the force field will determine how well the predictions are compared to experimental observables. Stacking in nucleic acids is one such structural property, originating mainly from London dispersion forces, which are quantum mechanical and are included in molecular mechanics force fields through nonbonded interactions. Geometric descriptions are utilized to decide if two residues are stacked and hence to calculate the stacking free energies for RNA dinucleoside monophosphates (DNMPs) through statistical mechanics for comparison with experimental thermodynamics data. Here, we benchmark four different stacking definitions using molecular dynamics (MD) trajectories for 16 RNA DNMPs produced by two different force fields (RNA-IL and ff99OL3) and show that our stacking definition better correlates with the experimental thermodynamics data. While predictions within an accuracy of 0.2 kcal/mol at 300 K were observed in RNA CC, CU, UC, AG, GA, and GG, stacked states of purine–pyrimidine and pyrimidine–purine DNMPs, respectively, were typically underpredicted and overpredicted. Additionally, population distributions of RNA UU DNMPs were poorly predicted by both force fields, implying a requirement for further force field revisions. We further discuss the differences predicted by each RNA force field. Finally, we show that discrete path sampling (DPS) calculations can provide valuable information and complement the MD simulations. We propose the use of experimental thermodynamics data for RNA DNMPs as benchmarks for testing RNA force fields.
中文翻译:

使用分子力学计算评估 RNA 二核苷单磷酸堆积的几何定义
通过小分子调节 RNA 是药物疗法中的一种新方法,其中确定 RNA 基序的结构特性被认为是开发能够靶向 RNA 结构以控制疾病的药物的有前途的方法。然而,由于 RNA 分子的复杂性和动态特性,使用实验方法确定 RNA 结构并不总是可行的,而采用力场的计算模型可以提供重要的见解。力场的质量将决定预测与实验观察结果相比的好坏程度。核酸中的堆积是一种这样的结构特性,主要源于伦敦色散力,它是量子力学的,通过非键相互作用包含在分子力学力场中。几何描述用于确定两个残基是否堆叠,从而通过统计力学计算 RNA 二核苷单磷酸 (DNMP) 的堆叠自由能,以便与实验热力学数据进行比较。在这里,我们使用分子动力学 (MD) 轨迹对由两个不同力场(RNA-IL 和 ff99OL3)产生的 16 个 RNA DNMP 的四种不同堆叠定义进行基准测试,并表明我们的堆叠定义与实验热力学数据更好地相关。虽然在 RNA CC、CU、UC、AG、GA 和 GG 中观察到在 300 K 时准确度为 0.2 kcal/mol 的预测,但嘌呤-嘧啶和嘧啶-嘌呤 DNMP 的堆叠状态通常分别被低估和高估。此外,RNA UU DNMP 的种群分布在两个力场中的预测都很差,这意味着需要进一步修正力场。我们进一步讨论了每个 RNA 力场预测的差异。最后,我们表明离散路径采样 (DPS) 计算可以提供有价值的信息并补充 MD 模拟。我们建议使用 RNA DNMP 的实验热力学数据作为测试 RNA 力场的基准。
更新日期:2022-06-02
中文翻译:
使用分子力学计算评估 RNA 二核苷单磷酸堆积的几何定义
通过小分子调节 RNA 是药物疗法中的一种新方法,其中确定 RNA 基序的结构特性被认为是开发能够靶向 RNA 结构以控制疾病的药物的有前途的方法。然而,由于 RNA 分子的复杂性和动态特性,使用实验方法确定 RNA 结构并不总是可行的,而采用力场的计算模型可以提供重要的见解。力场的质量将决定预测与实验观察结果相比的好坏程度。核酸中的堆积是一种这样的结构特性,主要源于伦敦色散力,它是量子力学的,通过非键相互作用包含在分子力学力场中。几何描述用于确定两个残基是否堆叠,从而通过统计力学计算 RNA 二核苷单磷酸 (DNMP) 的堆叠自由能,以便与实验热力学数据进行比较。在这里,我们使用分子动力学 (MD) 轨迹对由两个不同力场(RNA-IL 和 ff99OL3)产生的 16 个 RNA DNMP 的四种不同堆叠定义进行基准测试,并表明我们的堆叠定义与实验热力学数据更好地相关。虽然在 RNA CC、CU、UC、AG、GA 和 GG 中观察到在 300 K 时准确度为 0.2 kcal/mol 的预测,但嘌呤-嘧啶和嘧啶-嘌呤 DNMP 的堆叠状态通常分别被低估和高估。此外,RNA UU DNMP 的种群分布在两个力场中的预测都很差,这意味着需要进一步修正力场。我们进一步讨论了每个 RNA 力场预测的差异。最后,我们表明离散路径采样 (DPS) 计算可以提供有价值的信息并补充 MD 模拟。我们建议使用 RNA DNMP 的实验热力学数据作为测试 RNA 力场的基准。










































 京公网安备 11010802027423号
京公网安备 11010802027423号