Structure ( IF 4.4 ) Pub Date : 2022-05-20 , DOI: 10.1016/j.str.2022.04.013 Davide Sala 1 , Diego Del Alamo 2 , Hassane S Mchaourab 3 , Jens Meiler 4
|
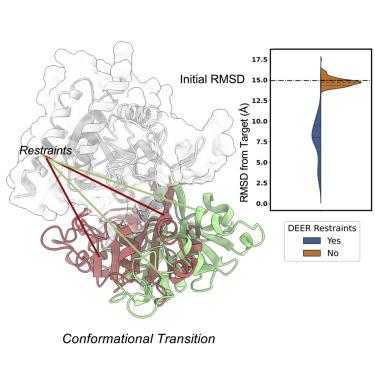
Conformational changes are an essential component of functional cycles of many proteins, but their characterization often requires an integrative structural biology approach. Here, we introduce and benchmark ConfChangeMover (CCM), a new method built into the widely used macromolecular modeling suite Rosetta that is tailored to model conformational changes in proteins using sparse experimental data. CCM can rotate and translate secondary structural elements and modify their backbone dihedral angles in regions of interest. We benchmarked CCM on soluble and membrane proteins with simulated Cα-Cα distance restraints and sparse experimental double electron-electron resonance (DEER) restraints, respectively. In both benchmarks, CCM outperformed state-of-the-art Rosetta methods, showing that it can model a diverse array of conformational changes. In addition, the Rosetta framework allows a wide variety of experimental data to be integrated with CCM, thus extending its capability beyond DEER restraints. This method will contribute to the biophysical characterization of protein dynamics.
中文翻译:
在有限实验数据的指导下使用 Rosetta 进行蛋白质构象变化建模
构象变化是许多蛋白质功能循环的重要组成部分,但它们的表征通常需要综合的结构生物学方法。在这里,我们介绍并基准测试 ConfChangeMover (CCM),这是一种内置于广泛使用的大分子建模套件 Rosetta 中的新方法,专门用于使用稀疏实验数据对蛋白质的构象变化进行建模。 CCM 可以旋转和平移二级结构元素,并修改感兴趣区域中的主干二面角。我们分别使用模拟 Cα-Cα 距离限制和稀疏实验双电子共振 (DEER) 限制对可溶性蛋白和膜蛋白的 CCM 进行基准测试。在这两个基准测试中,CCM 均优于最先进的 Rosetta 方法,表明它可以模拟多种构象变化。此外,Rosetta 框架允许将各种实验数据与 CCM 集成,从而将其功能扩展到 DEER 之外。该方法将有助于蛋白质动力学的生物物理表征。










































 京公网安备 11010802027423号
京公网安备 11010802027423号