当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Kinematic Vibrational Entropy Assessment and Analysis of SARS CoV-2 Main Protease
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-20 , DOI: 10.1021/acs.jcim.2c00126 Xiyu Chen 1 , Sigrid Leyendecker 1 , Henry van den Bedem 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-20 , DOI: 10.1021/acs.jcim.2c00126 Xiyu Chen 1 , Sigrid Leyendecker 1 , Henry van den Bedem 2
Affiliation
|
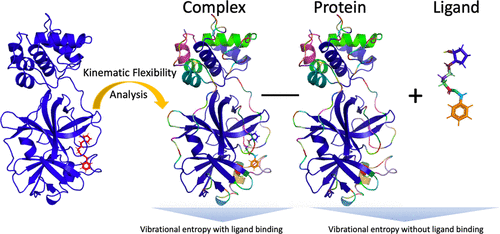
The three-dimensional conformations of a protein influence its function and select for the ligands it can interact with. The total free energy change during protein–ligand complex formation includes enthalphic and entropic components, which together report on the binding affinity and conformational states of the complex. However, determining the entropic contribution is computationally burdensome. Here, we apply kinematic flexibility analysis (KFA) to efficiently estimate vibrational frequencies from static protein and protein–ligand structures. The vibrational frequencies, in turn, determine the vibrational entropies of the structures and their complexes. Our estimates of the vibrational entropy change caused by ligand binding compare favorably to values obtained from a dynamic Normal Mode Analysis (NMA). Higher correlation factors can be achieved by increasing the distance cutoff in the potential energy model. Furthermore, we apply our new method to analyze the entropy changes of the SARS CoV-2 main protease when binding with different ligand inhibitors, which is relevant for the design of potential drugs.
中文翻译:

SARS CoV-2主要蛋白酶的运动学振动熵评估与分析
蛋白质的三维构象会影响其功能并选择可以与之相互作用的配体。蛋白质-配体复合物形成过程中的总自由能变化包括焓和熵成分,它们共同报告复合物的结合亲和力和构象状态。然而,确定熵贡献在计算上是繁重的。在这里,我们应用运动灵活性分析 (KFA) 来有效地估计静态蛋白质和蛋白质-配体结构的振动频率。反过来,振动频率决定了结构及其复合物的振动熵。我们对由配体结合引起的振动熵变化的估计与从动态正常模式分析 (NMA) 获得的值相比是有利的。通过增加势能模型中的距离截止可以实现更高的相关因子。此外,我们应用我们的新方法分析了 SARS CoV-2 主要蛋白酶与不同配体抑制剂结合时的熵变化,这与潜在药物的设计有关。
更新日期:2022-05-20
中文翻译:
SARS CoV-2主要蛋白酶的运动学振动熵评估与分析
蛋白质的三维构象会影响其功能并选择可以与之相互作用的配体。蛋白质-配体复合物形成过程中的总自由能变化包括焓和熵成分,它们共同报告复合物的结合亲和力和构象状态。然而,确定熵贡献在计算上是繁重的。在这里,我们应用运动灵活性分析 (KFA) 来有效地估计静态蛋白质和蛋白质-配体结构的振动频率。反过来,振动频率决定了结构及其复合物的振动熵。我们对由配体结合引起的振动熵变化的估计与从动态正常模式分析 (NMA) 获得的值相比是有利的。通过增加势能模型中的距离截止可以实现更高的相关因子。此外,我们应用我们的新方法分析了 SARS CoV-2 主要蛋白酶与不同配体抑制剂结合时的熵变化,这与潜在药物的设计有关。










































 京公网安备 11010802027423号
京公网安备 11010802027423号