当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Quantum chemical rovibrational analysis of aminoborane and its isotopologues
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2022-05-18 , DOI: 10.1002/jcc.26893 Moritz Schneider 1 , Guntram Rauhut 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2022-05-18 , DOI: 10.1002/jcc.26893 Moritz Schneider 1 , Guntram Rauhut 1
Affiliation
|
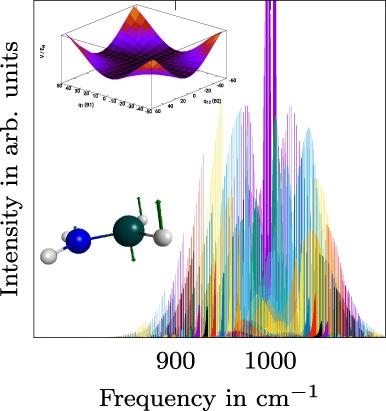
Aminoborane, H2NBH2 and its isotopologues, H2N10BH2, D2NBD2, and D2N10BD2, have been studied by high-level ab initio methods. All calculations rely on multidimensional potential energy surfaces and dipole moment surfaces including high-order mode coupling terms, which have been obtained from electronic structure calculations at the level of explicitly correlated coupled-cluster theory, CCSD(T)-F12, or the distinguishable cluster approximation, DCSD. Subsequent vibrational structure calculations based on second-order vibrational perturbation theory, VPT2, and vibrational configuration interaction theory, VCI, were used to determine rotational constants, centrifugal distortion constants, vibrationally averaged geometrical parameters and (an)harmonic vibrational frequencies. The impact of core-correlation effects is discussed in detail. Rovibrational VCI calculations were used to simulate the gas phase spectra of these species and an in-depth analysis of the ν7 band of aminoborane is provided. Color-coding is used to reveal the identity of the individual progressions of the rovibrational transitions for this particular mode.
中文翻译:

氨基硼烷及其同位素体的量子化学振动分析
氨基硼烷 H 2 NBH 2及其同位素体 H 2 N 10 BH 2、D 2 NBD 2和 D 2 N 10 BD 2, 已通过高级从头算方法进行了研究。所有计算都依赖于多维势能面和偶极矩面,包括高阶模式耦合项,这些项是在显式相关耦合簇理论、CCSD(T)-F12 或可区分簇的水平上从电子结构计算中获得的近似值,DCSD。基于二阶振动微扰理论 VPT2 和振动构型相互作用理论 VCI 的后续振动结构计算用于确定旋转常数、离心畸变常数、振动平均几何参数和(谐波)振动频率。详细讨论了核心相关效应的影响。提供了氨基硼烷的ν 7谱带。颜色编码用于揭示此特定模式的振动过渡的各个进程的身份。
更新日期:2022-05-18
中文翻译:
氨基硼烷及其同位素体的量子化学振动分析
氨基硼烷 H 2 NBH 2及其同位素体 H 2 N 10 BH 2、D 2 NBD 2和 D 2 N 10 BD 2, 已通过高级从头算方法进行了研究。所有计算都依赖于多维势能面和偶极矩面,包括高阶模式耦合项,这些项是在显式相关耦合簇理论、CCSD(T)-F12 或可区分簇的水平上从电子结构计算中获得的近似值,DCSD。基于二阶振动微扰理论 VPT2 和振动构型相互作用理论 VCI 的后续振动结构计算用于确定旋转常数、离心畸变常数、振动平均几何参数和(谐波)振动频率。详细讨论了核心相关效应的影响。提供了氨基硼烷的ν 7谱带。颜色编码用于揭示此特定模式的振动过渡的各个进程的身份。


























 京公网安备 11010802027423号
京公网安备 11010802027423号