当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanistic Modeling of Monoglyceride Lipase Covalent Modification Elucidates the Role of Leaving Group Expulsion and Discriminates Inhibitors with High and Low Potency
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-17 , DOI: 10.1021/acs.jcim.2c00140 Francesca Galvani 1 , Laura Scalvini 1 , Silvia Rivara 1 , Alessio Lodola 1 , Marco Mor 1, 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-17 , DOI: 10.1021/acs.jcim.2c00140 Francesca Galvani 1 , Laura Scalvini 1 , Silvia Rivara 1 , Alessio Lodola 1 , Marco Mor 1, 2
Affiliation
|
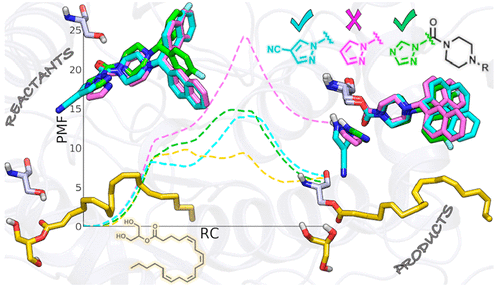
Inhibition of monoglyceride lipase (MGL), also known as monoacylglycerol lipase (MAGL), has emerged as a promising approach for treating neurological diseases. To gain useful insights in the design of agents with balanced potency and reactivity, we investigated the mechanism of MGL carbamoylation by the reference triazole urea SAR629 (IC50 = 0.2 nM) and two recently described inhibitors featuring a pyrazole (IC50 = 1800 nM) or a 4-cyanopyrazole (IC50 = 8 nM) leaving group (LG), using a hybrid quantum mechanics/molecular mechanics (QM/MM) approach. Opposite to what was found for substrate 2-arachidonoyl-sn-glycerol (2-AG), covalent modification of MGL by azole ureas is controlled by LG expulsion. Simulations indicated that changes in the electronic structure of the LG greatly affect reaction energetics with triazole and 4-cyanopyrazole inhibitors following a more accessible carbamoylation path compared to the unsubstituted pyrazole derivative. The computational protocol provided reaction barriers able to discriminate between MGL inhibitors with different potencies. These results highlight how QM/MM simulations can contribute to elucidating structure–activity relationships and provide insights for the design of covalent inhibitors.
中文翻译:

单甘酯脂肪酶共价修饰的机械模型阐明了离去基团排斥的作用并区分了高效和低效的抑制剂
抑制甘油单酯脂肪酶 (MGL),也称为单酰基甘油脂肪酶 (MAGL),已成为治疗神经系统疾病的有前景的方法。为了在设计具有平衡效力和反应性的药剂时获得有用的见解,我们通过参考三唑脲 SAR629 (IC 50 = 0.2 nM) 和最近描述的两种吡唑抑制剂 (IC 50 = 1800 nM)研究了 MGL 氨基甲酰化的机制或 4-氰基吡唑 (IC 50 = 8 nM) 离去基团 (LG),使用混合量子力学/分子力学 (QM/MM) 方法。与底物 2-花生四烯酰-sn的发现相反-甘油 (2-AG),唑脲对 MGL 的共价修饰受 LG 排出控制。模拟表明,与未取代的吡唑衍生物相比,LG 电子结构的变化极大地影响了与三唑和 4-氰基吡唑抑制剂的反应能量学,其氨基甲酰化途径更容易获得。计算方案提供了能够区分具有不同效力的 MGL 抑制剂的反应障碍。这些结果突出了 QM/MM 模拟如何有助于阐明构效关系并为共价抑制剂的设计提供见解。
更新日期:2022-05-17
中文翻译:
单甘酯脂肪酶共价修饰的机械模型阐明了离去基团排斥的作用并区分了高效和低效的抑制剂
抑制甘油单酯脂肪酶 (MGL),也称为单酰基甘油脂肪酶 (MAGL),已成为治疗神经系统疾病的有前景的方法。为了在设计具有平衡效力和反应性的药剂时获得有用的见解,我们通过参考三唑脲 SAR629 (IC 50 = 0.2 nM) 和最近描述的两种吡唑抑制剂 (IC 50 = 1800 nM)研究了 MGL 氨基甲酰化的机制或 4-氰基吡唑 (IC 50 = 8 nM) 离去基团 (LG),使用混合量子力学/分子力学 (QM/MM) 方法。与底物 2-花生四烯酰-sn的发现相反-甘油 (2-AG),唑脲对 MGL 的共价修饰受 LG 排出控制。模拟表明,与未取代的吡唑衍生物相比,LG 电子结构的变化极大地影响了与三唑和 4-氰基吡唑抑制剂的反应能量学,其氨基甲酰化途径更容易获得。计算方案提供了能够区分具有不同效力的 MGL 抑制剂的反应障碍。这些结果突出了 QM/MM 模拟如何有助于阐明构效关系并为共价抑制剂的设计提供见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号