当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Transferable Classical Force Field for Pure and Mixed Metal Halide Perovskites Parameterized from First-Principles
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-16 , DOI: 10.1021/acs.jcim.1c01506 Juan Antonio Seijas-Bellido 1 , Bipasa Samanta 2 , Karen Valadez-Villalobos 1 , Juan Jesús Gallardo 3 , Javier Navas 3 , Salvador R G Balestra 1, 4 , Rafael María Madero Castro 1 , José Manuel Vicent-Luna 5 , Shuxia Tao 5 , Maytal Caspary Toroker 2 , Juan Antonio Anta 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-16 , DOI: 10.1021/acs.jcim.1c01506 Juan Antonio Seijas-Bellido 1 , Bipasa Samanta 2 , Karen Valadez-Villalobos 1 , Juan Jesús Gallardo 3 , Javier Navas 3 , Salvador R G Balestra 1, 4 , Rafael María Madero Castro 1 , José Manuel Vicent-Luna 5 , Shuxia Tao 5 , Maytal Caspary Toroker 2 , Juan Antonio Anta 1
Affiliation
|
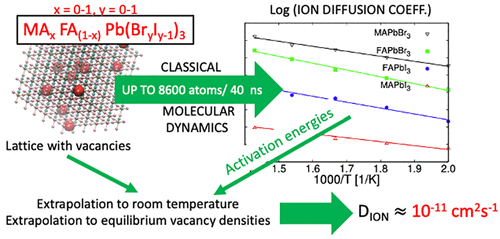
Many key features in photovoltaic perovskites occur in relatively long time scales and involve mixed compositions. This requires realistic but also numerically simple models. In this work we present a transferable classical force field to describe the mixed hybrid perovskite MAxFA1–xPb(BryI1–y)3 for variable composition (∀x, y ∈ [0, 1]). The model includes Lennard-Jones and Buckingham potentials to describe the interactions between the atoms of the inorganic lattice and the organic molecule, and the AMBER model to describe intramolecular atomic interactions. Most of the parameters of the force field have been obtained by means of a genetic algorithm previously developed to parametrize the CsPb(BrxI1–x)3 perovskite (Balestra et al. J. Mater. Chem. A. 2020, DOI: 10.1039/d0ta03200j). The algorithm finds the best parameter set that simultaneously fits the DFT energies obtained for several crystalline structures with moderate degrees of distortion with respect to the equilibrium configuration. The resulting model reproduces correctly the XRD patterns, the expansion of the lattice upon I/Br substitution, and the thermal expansion coefficients. We use the model to run classical molecular dynamics simulations with up to 8600 atoms and simulation times of up to 40 ns. From the simulations we have extracted the ion diffusion coefficient of the pure and mixed perovskites, presenting for the first time these values obtained by a fully dynamical method using a transferable model fitted to first-principles calculations. The values here reported can be considered as the theoretical upper limit, that is, without grain boundaries or other defects, for ion migration dynamics induced by halide vacancies in photovoltaic perovskite devices under operational conditions.
中文翻译:

根据第一原理参数化的纯金属卤化物钙钛矿和混合金属卤化物钙钛矿的可传递经典力场
光伏钙钛矿的许多关键特征发生在相对较长的时间范围内,并且涉及混合成分。这需要现实且数值简单的模型。在这项工作中,我们提出了一个可转移的经典力场来描述混合杂化钙钛矿 MA x FA 1– x Pb(Br y I 1– y ) 3的可变成分 (∀ x , y ∈ [0, 1])。该模型包括用于描述无机晶格原子与有机分子之间相互作用的 Lennard-Jones 和 Buckingham 势,以及用于描述分子内原子相互作用的 AMBER 模型。力场的大部分参数都是通过先前开发的用于参数化 CsPb(Br x I 1– x ) 3钙钛矿的遗传算法获得的(Balestra 等人J. Mater. Chem. A . 2020 ,DOI: 10.1039/d0ta03200j)。该算法找到了最佳参数集,该参数集同时拟合了几种晶体结构获得的 DFT 能量,这些晶体结构相对于平衡构型具有中等程度的畸变。所得模型正确再现了 XRD 图案、I/Br 取代时晶格的膨胀以及热膨胀系数。我们使用该模型运行多达 8600 个原子的经典分子动力学模拟,模拟时间长达 40 ns。从模拟中,我们提取了纯钙钛矿和混合钙钛矿的离子扩散系数,首次展示了使用适合第一原理计算的可转移模型通过全动态方法获得的这些值。 这里报告的值可以被认为是操作条件下光伏钙钛矿器件中卤化物空位引起的离子迁移动力学的理论上限,即没有晶界或其他缺陷。
更新日期:2022-05-16
中文翻译:
根据第一原理参数化的纯金属卤化物钙钛矿和混合金属卤化物钙钛矿的可传递经典力场
光伏钙钛矿的许多关键特征发生在相对较长的时间范围内,并且涉及混合成分。这需要现实且数值简单的模型。在这项工作中,我们提出了一个可转移的经典力场来描述混合杂化钙钛矿 MA x FA 1– x Pb(Br y I 1– y ) 3的可变成分 (∀ x , y ∈ [0, 1])。该模型包括用于描述无机晶格原子与有机分子之间相互作用的 Lennard-Jones 和 Buckingham 势,以及用于描述分子内原子相互作用的 AMBER 模型。力场的大部分参数都是通过先前开发的用于参数化 CsPb(Br x I 1– x ) 3钙钛矿的遗传算法获得的(Balestra 等人J. Mater. Chem. A . 2020 ,DOI: 10.1039/d0ta03200j)。该算法找到了最佳参数集,该参数集同时拟合了几种晶体结构获得的 DFT 能量,这些晶体结构相对于平衡构型具有中等程度的畸变。所得模型正确再现了 XRD 图案、I/Br 取代时晶格的膨胀以及热膨胀系数。我们使用该模型运行多达 8600 个原子的经典分子动力学模拟,模拟时间长达 40 ns。从模拟中,我们提取了纯钙钛矿和混合钙钛矿的离子扩散系数,首次展示了使用适合第一原理计算的可转移模型通过全动态方法获得的这些值。 这里报告的值可以被认为是操作条件下光伏钙钛矿器件中卤化物空位引起的离子迁移动力学的理论上限,即没有晶界或其他缺陷。










































 京公网安备 11010802027423号
京公网安备 11010802027423号