当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Development of a Hybrid-Resolution Force Field for Peptide Self-Assembly Simulations: Optimizing Peptide–Peptide and Peptide–Solvent Interactions
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-13 , DOI: 10.1021/acs.jcim.2c00066 Xiang Cai 1 , Wei Han 1, 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-05-13 , DOI: 10.1021/acs.jcim.2c00066 Xiang Cai 1 , Wei Han 1, 2
Affiliation
|
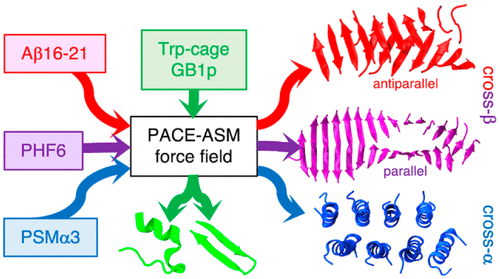
Atomic descriptions of peptide self-assembly are crucial to an understanding of disease-related peptide aggregation and the design of peptide-assembled materials. Obtaining these descriptions through computer simulation is challenging because current force fields, which were not designed for this process and are often unable to describe correctly peptide self-assembly behavior and the sequence dependence. Here, we developed a framework using dipeptide aggregation as a model system to improve force fields for simulations of self-assembly. Aggregation-related structural properties were designed and used to guide the optimization of peptide–peptide and peptide–solvent interactions. With this framework, we developed a self-assembly force field, termed PACE-ASM, by reoptimizing a hybrid-resolution force field that was originally developed for folding simulation. With its applicability in folding simulations, the new PACE was used to simulate the self-assembly of two disease-related short peptides, Aβ16–21 and PHF6, into β-sheet-rich cross-β amyloids. These simulations reproduced the crystal structures of Aβ16–21 and PHF6 amyloids at near-atomic resolution and captured the difference in packing orientations between the two sequences, a task which is challenging even with all-atom force fields. Apart from cross-β amyloids, the self-assembly of emerging helix-rich cross-α amyloids by another peptide PSMα3 can also be correctly described with the new PACE, manifesting the versatility of the force field. We demonstrated that the ability of the PACE-ASM to model peptide self-assembly is based largely on its improved description of peptide–peptide and peptide–solvent interactions. This was achieved with our optimization framework that can readily identify and address the deficiency in describing these interactions.
中文翻译:

用于肽自组装模拟的混合分辨率力场的开发:优化肽-肽和肽-溶剂相互作用
肽自组装的原子描述对于理解与疾病相关的肽聚集和肽组装材料的设计至关重要。通过计算机模拟获得这些描述具有挑战性,因为当前的力场不是为此过程设计的,并且通常无法正确描述肽自组装行为和序列依赖性。在这里,我们开发了一个框架,使用二肽聚合作为模型系统来改善自组装模拟的力场。设计了与聚集相关的结构特性,并用于指导优化肽-肽和肽-溶剂相互作用。有了这个框架,我们开发了一个自组装力场,称为 PACE-ASM,通过重新优化最初为折叠模拟而开发的混合分辨率力场。凭借其在折叠模拟中的适用性,新的 PACE 被用于模拟两种与疾病相关的短肽 Aβ 的自组装16-21和 PHF6,转化为富含 β-折叠的交叉-β 淀粉样蛋白。这些模拟再现了 Aβ 16-21的晶体结构和 PHF6 淀粉样蛋白以近原子分辨率捕获并捕获了两个序列之间包装方向的差异,即使使用全原子力场也是一项具有挑战性的任务。除了交叉β淀粉样蛋白外,另一种肽PSMα3对新兴的富含螺旋的交叉α淀粉样蛋白的自组装也可以用新的PACE正确描述,体现了力场的多功能性。我们证明了 PACE-ASM 模拟肽自组装的能力主要基于其对肽-肽和肽-溶剂相互作用的改进描述。这是通过我们的优化框架实现的,该框架可以轻松识别和解决描述这些交互的缺陷。
更新日期:2022-05-13
中文翻译:
用于肽自组装模拟的混合分辨率力场的开发:优化肽-肽和肽-溶剂相互作用
肽自组装的原子描述对于理解与疾病相关的肽聚集和肽组装材料的设计至关重要。通过计算机模拟获得这些描述具有挑战性,因为当前的力场不是为此过程设计的,并且通常无法正确描述肽自组装行为和序列依赖性。在这里,我们开发了一个框架,使用二肽聚合作为模型系统来改善自组装模拟的力场。设计了与聚集相关的结构特性,并用于指导优化肽-肽和肽-溶剂相互作用。有了这个框架,我们开发了一个自组装力场,称为 PACE-ASM,通过重新优化最初为折叠模拟而开发的混合分辨率力场。凭借其在折叠模拟中的适用性,新的 PACE 被用于模拟两种与疾病相关的短肽 Aβ 的自组装16-21和 PHF6,转化为富含 β-折叠的交叉-β 淀粉样蛋白。这些模拟再现了 Aβ 16-21的晶体结构和 PHF6 淀粉样蛋白以近原子分辨率捕获并捕获了两个序列之间包装方向的差异,即使使用全原子力场也是一项具有挑战性的任务。除了交叉β淀粉样蛋白外,另一种肽PSMα3对新兴的富含螺旋的交叉α淀粉样蛋白的自组装也可以用新的PACE正确描述,体现了力场的多功能性。我们证明了 PACE-ASM 模拟肽自组装的能力主要基于其对肽-肽和肽-溶剂相互作用的改进描述。这是通过我们的优化框架实现的,该框架可以轻松识别和解决描述这些交互的缺陷。










































 京公网安备 11010802027423号
京公网安备 11010802027423号