当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Investigation of water substitution at RuII complexes by conceptual density function theory approach
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-04-29 , DOI: 10.1002/jcc.26878 Dušan Ćoćić 1 , Biljana Petrović 1 , Ralph Puchta 2, 3, 4 , Marta Chrzanowska 5 , Anna Katafias 5 , Rudi van Eldik 2, 5
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-04-29 , DOI: 10.1002/jcc.26878 Dušan Ćoćić 1 , Biljana Petrović 1 , Ralph Puchta 2, 3, 4 , Marta Chrzanowska 5 , Anna Katafias 5 , Rudi van Eldik 2, 5
Affiliation
|
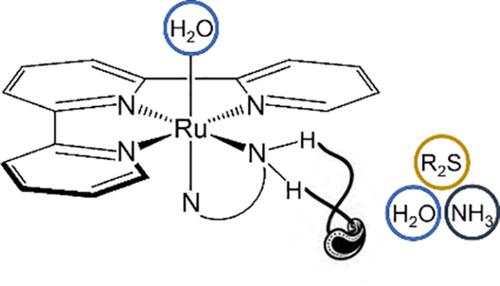
In this paper, we investigated water exchange reactions and substitution of aqua RuII complexes of general formula [Ru(terpy)(N^N)(H2O)]2+ (where N^N = ethylenediamine (en), 1,2-(aminomethyl)pyridine (ampy) and 2,2′-bipyridine (bipy)) by ammonia and thioformaldehyde. These reactions were studied in detail by applying conceptual density functional theory. This approach enabled us to gain further insight into the underlying reaction mechanism at the microscopic level (involving only direct participants of the reaction, without the influence of the solvent) and to put the concept of reaction mechanism on a quantitative basis. The course of the chemical reaction along the reaction coordinate ξ, is rationalized in terms of reaction energy, force, dipole moment, and reaction electronic flux (REF). The results yield and characterize the significant influence of an intermolecular hydrogen bond formed between the entering and the spectator ligand to the overall energy barrier of the reactions.
中文翻译:

用概念密度函数理论方法研究 RuII 配合物的水替代
在本文中,我们研究了通式 [Ru(terpy)(N^N)(H 2 O )] 2+ (其中N ^ N = 乙二胺 (en), 1, 2-(氨基甲基)吡啶 (ampy) 和 2,2'-联吡啶 (bipy)) 由氨和硫代甲醛生成。通过应用概念密度泛函理论对这些反应进行了详细研究。这种方法使我们能够在微观层面进一步了解潜在的反应机制(仅涉及反应的直接参与者,不受溶剂的影响),并将反应机制的概念置于定量的基础上。沿反应坐标ξ的化学反应过程, 在反应能量、力、偶极矩和反应电子通量 (REF) 方面进行了合理化。结果产生并表征了在进入配体和旁观配体之间形成的分子间氢键对反应的整体能垒的显着影响。
更新日期:2022-04-29
中文翻译:
用概念密度函数理论方法研究 RuII 配合物的水替代
在本文中,我们研究了通式 [Ru(terpy)(N^N)(H 2 O )] 2+ (其中N ^ N = 乙二胺 (en), 1, 2-(氨基甲基)吡啶 (ampy) 和 2,2'-联吡啶 (bipy)) 由氨和硫代甲醛生成。通过应用概念密度泛函理论对这些反应进行了详细研究。这种方法使我们能够在微观层面进一步了解潜在的反应机制(仅涉及反应的直接参与者,不受溶剂的影响),并将反应机制的概念置于定量的基础上。沿反应坐标ξ的化学反应过程, 在反应能量、力、偶极矩和反应电子通量 (REF) 方面进行了合理化。结果产生并表征了在进入配体和旁观配体之间形成的分子间氢键对反应的整体能垒的显着影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号