Solid State Nuclear Magnetic Resonance ( IF 3.2 ) Pub Date : 2022-04-28 , DOI: 10.1016/j.ssnmr.2022.101795 Scott A Southern 1 , David L Bryce 1
|
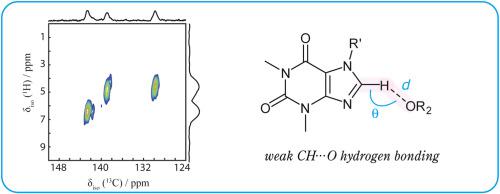
Weak hydrogen bonds are important structure-directing elements in supramolecular chemistry and biochemistry. We consider here weak CH⋯O hydrogen bonds in a series of cocrystals of theophylline and caffeine and assess to what extent the CH⋯O distance and angle govern the observed 13C and 1H isotropic chemical shifts. Gauge-including projector-augmented wave density functional theory (GIPAW DFT) calculations consistently predict a decrease in the 13C and 1H magnetic shielding constants upon hydrogen bond formation on the order of 2–5 ppm (13C) and 1–2 ppm (1H). These trends are reproduced using the machine-learning approach implemented in ShiftML. Experimental 13C and 1H chemical shifts obtained for powdered samples using one-dimensional NMR spectroscopy as well as heteronuclear correlation (HETCOR) spectroscopy correlate well with the GIPAW DFT results. However, the experimental 13C NMR response only correlates moderately well with the hydrogen bond length and angle, while the experimental 1H chemical shifts only show very weak correlations to these local structural elements. DFT computations on isolated imidazole-formaldehyde models show that the 13C and 1H chemical shifts generally decrease with the C⋯O distance but show no clear dependence on the CH⋯O angle. These results demonstrate that the 13C and 1H response to weak CH⋯O hydrogen bonding is influenced significantly by additional weak contacts within cocrystal heterodimeric units.
中文翻译:
键长和键角在多大程度上控制 13C 和 1H NMR 对弱 CH⋯O 氢键的响应?咖啡因和茶碱共晶体的案例研究
弱氢键是超分子化学和生物化学中重要的结构导向元素。我们在这里考虑茶碱和咖啡因的一系列共晶体中的弱 CH⋯O 氢键,并评估 CH⋯O 距离和角度在多大程度上控制观察到的13 C 和1 H 各向同性化学位移。包括投影仪的仪表增强波密度泛函理论 (GIPAW DFT) 计算一致地预测13 C 和1 H 磁屏蔽常数在氢键形成时降低约 2-5 ppm ( 13 C) 和 1-2 ppm ( 1小时)。使用 ShiftML 中实现的机器学习方法再现了这些趋势。实验13使用一维 NMR 光谱以及异核相关 (HETCOR) 光谱获得的粉末样品的C 和1 H 化学位移与 GIPAW DFT 结果具有很好的相关性。然而,实验13 C NMR 响应仅与氢键长度和角度有适度的相关性,而实验1 H 化学位移仅显示与这些局部结构元素的非常弱的相关性。对孤立的咪唑-甲醛模型的 DFT 计算表明,13 C 和1 H 化学位移通常随着 C⋯O 距离的增加而减小,但对 CH⋯O 角没有明显的依赖性。这些结果表明13 C 和1H对弱CH⋯O氢键的响应受到共晶异二聚体单元内额外的弱接触的显着影响。


























 京公网安备 11010802027423号
京公网安备 11010802027423号