Journal of Advanced Research ( IF 10.7 ) Pub Date : 2022-04-12 , DOI: 10.1016/j.jare.2022.04.004 Mária Škrabišová 1 , Nicholas Dietz 2 , Shuai Zeng 3 , Yen On Chan 4 , Juexin Wang 3 , Yang Liu 4 , Jana Biová 1 , Trupti Joshi 5 , Kristin D Bilyeu 6
|
Introduction
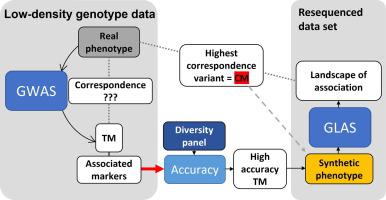
Genome-Wide Association Studies (GWAS) identify tagging variants in the genome that are statistically associated with the phenotype because of their linkage disequilibrium (LD) relationship with the causative mutation (CM). When both low-density genotyped accession panels with phenotypes and resequenced data accession panels are available, tagging variants can assist with post-GWAS challenges in CM discovery.
Objectives
Our objective was to identify additional GWAS evaluation criteria to assess correspondence between genomic variants and phenotypes, as well as enable deeper analysis of the localized landscape of association.
Methods
We used genomic variant positions as Synthetic phenotypes in GWAS that we named “Synthetic phenotype association study” (SPAS). The extreme case of SPAS is what we call an “Inverse GWAS” where we used CM positions of cloned soybean genes. We developed and validated the Accuracy concept as a measure of the correspondence between variant positions and phenotypes.
Results
The SPAS approach demonstrated that the genotype status of an associated variant used as a Synthetic phenotype enabled us to explore the relationships between tagging variants and CMs, and further, that utilizing CMs as Synthetic phenotypes in Inverse GWAS illuminated the landscape of association. We implemented the Accuracy calculation for a curated accession panel to an online Accuracy calculation tool (AccuTool) as a resource for gene identification in soybean. We demonstrated our concepts on three examples of soybean cloned genes. As a result of our findings, we devised an enhanced “GWAS to Genes” analysis (Synthetic phenotype to CM strategy, SP2CM). Using SP2CM, we identified a CM for a novel gene.
Conclusion
The SP2CM strategy utilizing Synthetic phenotypes and the Accuracy calculation of correspondence provides crucial information to assist researchers in CM discovery. The impact of this work is a more effective evaluation of landscapes of GWAS associations.
中文翻译:
一种新的合成表型关联研究方法揭示了基因组变异和表型的关联景观
介绍
全基因组关联研究 (GWAS) 识别基因组中与表型统计相关的标记变体,因为它们与致病突变 (CM) 存在连锁不平衡 (LD) 关系。当具有表型的低密度基因分型登录面板和重新排序的数据登录面板都可用时,标记变体可以帮助解决 CM 发现中的后 GWAS 挑战。
目标
我们的目标是确定额外的 GWAS 评估标准,以评估基因组变异和表型之间的对应关系,以及对局部关联景观进行更深入的分析。
方法
我们使用基因组变异位置作为 GWAS 中的合成表型,我们将其命名为“合成表型关联研究”(SPAS)。SPAS 的极端情况就是我们所说的“反向 GWAS”,我们使用克隆大豆基因的 CM 位置。我们开发并验证了准确性概念作为变异位置和表型之间对应关系的度量。
结果
SPAS 方法表明,用作合成表型的相关变体的基因型状态使我们能够探索标记变体和 CM 之间的关系,并且进一步地,利用 CM 作为逆 GWAS 中的合成表型阐明了关联的景观。我们对在线准确度计算工具 (AccuTool) 进行了精选登录面板的准确度计算,作为大豆基因鉴定的资源。我们在大豆克隆基因的三个例子中展示了我们的概念。作为我们发现的结果,我们设计了一种增强的“GWAS 到基因”分析(合成表型到 CM 策略,SP2CM)。使用 SP2CM,我们确定了一个新基因的 CM。
结论
利用合成表型的 SP2CM 策略和对应的准确度计算为协助研究人员进行 CM 发现提供了重要信息。这项工作的影响是更有效地评估 GWAS 协会的景观。


























 京公网安备 11010802027423号
京公网安备 11010802027423号