当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Sequential design of adsorption simulations in metal–organic frameworks
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2021-12-09 , DOI: 10.1039/d1me00138h Krishnendu Mukherjee 1 , Alexander W. Dowling 1 , Yamil J. Colón 1
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2021-12-09 , DOI: 10.1039/d1me00138h Krishnendu Mukherjee 1 , Alexander W. Dowling 1 , Yamil J. Colón 1
Affiliation
|
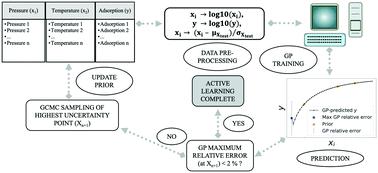
The large number of possible structures of metal–organic frameworks (MOFs) and their limitless potential applications have motivated molecular modelers and researchers to develop methods and models to efficiently assess MOF performance. Some of the techniques include large-scale high-throughput molecular simulations and machine learning models. Despite those advances, the number of possible materials and the potential conditions that could be used still pose a formidable challenge for model development requiring large data sets. Therefore, there is a clear need for algorithms that can efficiently explore the spaces while balancing the number of simulations with prediction accuracy. Here, we present how active learning can sequentially select simulation conditions for gas adsorption, ultimately resulting in accurate adsorption predictions with an order of magnitude lower number of simulations. We model adsorption of pure components methane and carbon dioxide in Cu–BTC. We employ Gaussian process regression (GPR) and use the resulting uncertainties in the predictions to guide the next sampling point for molecular simulation. We outline the procedure and demonstrate how this model can emulate adsorption isotherms at 300 K from 10−6 to 300 bar (methane)/100 bar (carbon dioxide). We also show how this procedure can be used for predicting adsorption on a temperature–pressure phase space for a temperature range of 100 to 300 K, and pressure range of 10−6 to 300 bar (methane)/100 bar (carbon dioxide).
中文翻译:

金属-有机骨架中吸附模拟的顺序设计
金属有机骨架 (MOF) 的大量可能结构及其无限的潜在应用促使分子建模者和研究人员开发方法和模型来有效评估 MOF 的性能。其中一些技术包括大规模高通量分子模拟和机器学习模型。尽管取得了这些进展,但可能使用的材料数量和潜在条件仍然对需要大数据集的模型开发构成了巨大挑战。因此,显然需要能够有效探索空间同时平衡模拟数量与预测精度的算法。在这里,我们介绍了主动学习如何依次选择气体吸附的模拟条件,最终导致准确的吸附预测,模拟次数减少一个数量级。我们模拟了 Cu-BTC 中纯组分甲烷和二氧化碳的吸附。我们采用高斯过程回归 (GPR) 并使用预测中产生的不确定性来指导分子模拟的下一个采样点。我们概述了该过程并演示了该模型如何模拟 300 K 下的吸附等温线,从 10−6至 300 巴(甲烷)/100 巴(二氧化碳)。我们还展示了该程序如何用于预测温度范围为 100 至 300 K 和压力范围为 10 -6至 300 巴(甲烷)/100 巴(二氧化碳)的温度-压力相空间上的吸附。
更新日期:2021-12-16
中文翻译:
金属-有机骨架中吸附模拟的顺序设计
金属有机骨架 (MOF) 的大量可能结构及其无限的潜在应用促使分子建模者和研究人员开发方法和模型来有效评估 MOF 的性能。其中一些技术包括大规模高通量分子模拟和机器学习模型。尽管取得了这些进展,但可能使用的材料数量和潜在条件仍然对需要大数据集的模型开发构成了巨大挑战。因此,显然需要能够有效探索空间同时平衡模拟数量与预测精度的算法。在这里,我们介绍了主动学习如何依次选择气体吸附的模拟条件,最终导致准确的吸附预测,模拟次数减少一个数量级。我们模拟了 Cu-BTC 中纯组分甲烷和二氧化碳的吸附。我们采用高斯过程回归 (GPR) 并使用预测中产生的不确定性来指导分子模拟的下一个采样点。我们概述了该过程并演示了该模型如何模拟 300 K 下的吸附等温线,从 10−6至 300 巴(甲烷)/100 巴(二氧化碳)。我们还展示了该程序如何用于预测温度范围为 100 至 300 K 和压力范围为 10 -6至 300 巴(甲烷)/100 巴(二氧化碳)的温度-压力相空间上的吸附。










































 京公网安备 11010802027423号
京公网安备 11010802027423号