当前位置:
X-MOL 学术
›
Pharmacol. Rev.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure-Based Virtual Screening for Ligands of G Protein–Coupled Receptors: What Can Molecular Docking Do for You?
Pharmacological Reviews ( IF 19.3 ) Pub Date : 2021-10-01 , DOI: 10.1124/pharmrev.120.000246 Flavio Ballante 1 , Albert J Kooistra 1 , Stefanie Kampen 1 , Chris de Graaf 1 , Jens Carlsson 2
Pharmacological Reviews ( IF 19.3 ) Pub Date : 2021-10-01 , DOI: 10.1124/pharmrev.120.000246 Flavio Ballante 1 , Albert J Kooistra 1 , Stefanie Kampen 1 , Chris de Graaf 1 , Jens Carlsson 2
Affiliation
|
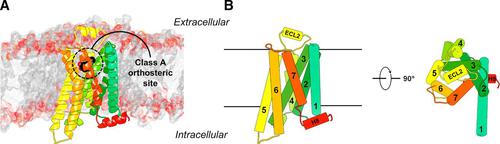
G protein–coupled receptors (GPCRs) constitute the largest family of membrane proteins in the human genome and are important therapeutic targets. During the last decade, the number of atomic-resolution structures of GPCRs has increased rapidly, providing insights into drug binding at the molecular level. These breakthroughs have created excitement regarding the potential of using structural information in ligand design and initiated a new era of rational drug discovery for GPCRs. The molecular docking method is now widely applied to model the three-dimensional structures of GPCR-ligand complexes and screen for chemical probes in large compound libraries. In this review article, we first summarize the current structural coverage of the GPCR superfamily and the understanding of receptor-ligand interactions at atomic resolution. We then present the general workflow of structure-based virtual screening and strategies to discover GPCR ligands in chemical libraries. We assess the state of the art of this research field by summarizing prospective applications of virtual screening based on experimental structures. Strategies to identify compounds with specific efficacy and selectivity profiles are discussed, illustrating the opportunities and limitations of the molecular docking method. Our overview shows that structure-based virtual screening can discover novel leads and will be essential in pursuing the next generation of GPCR drugs.
中文翻译:

基于结构的 G 蛋白偶联受体配体虚拟筛选:分子对接能为您做什么?
G 蛋白偶联受体 (GPCR) 构成人类基因组中最大的膜蛋白家族,是重要的治疗靶点。在过去十年中,GPCR 的原子分辨率结构的数量迅速增加,为分子水平的药物结合提供了见解。这些突破使人们对在配体设计中使用结构信息的潜力感到兴奋,并开启了 GPCR 合理药物发现的新时代。分子对接方法现在广泛应用于模拟 GPCR-配体复合物的三维结构和筛选大型化合物库中的化学探针。在这篇综述文章中,我们首先总结了 GPCR 超家族的当前结构覆盖范围以及对原子分辨率下受体-配体相互作用的理解。然后,我们介绍了基于结构的虚拟筛选的一般工作流程和在化学文库中发现 GPCR 配体的策略。我们通过总结基于实验结构的虚拟筛选的前瞻性应用来评估该研究领域的最新技术。讨论了识别具有特定功效和选择性特征的化合物的策略,说明了分子对接方法的机会和局限性。我们的概述表明,基于结构的虚拟筛选可以发现新的线索,并且对于追求下一代 GPCR 药物至关重要。我们通过总结基于实验结构的虚拟筛选的前瞻性应用来评估该研究领域的最新技术。讨论了识别具有特定功效和选择性特征的化合物的策略,说明了分子对接方法的机会和局限性。我们的概述表明,基于结构的虚拟筛选可以发现新的线索,并且对于追求下一代 GPCR 药物至关重要。我们通过总结基于实验结构的虚拟筛选的前瞻性应用来评估该研究领域的最新技术。讨论了识别具有特定功效和选择性特征的化合物的策略,说明了分子对接方法的机会和局限性。我们的概述表明,基于结构的虚拟筛选可以发现新的线索,并且对于追求下一代 GPCR 药物至关重要。
更新日期:2021-10-01
中文翻译:
基于结构的 G 蛋白偶联受体配体虚拟筛选:分子对接能为您做什么?
G 蛋白偶联受体 (GPCR) 构成人类基因组中最大的膜蛋白家族,是重要的治疗靶点。在过去十年中,GPCR 的原子分辨率结构的数量迅速增加,为分子水平的药物结合提供了见解。这些突破使人们对在配体设计中使用结构信息的潜力感到兴奋,并开启了 GPCR 合理药物发现的新时代。分子对接方法现在广泛应用于模拟 GPCR-配体复合物的三维结构和筛选大型化合物库中的化学探针。在这篇综述文章中,我们首先总结了 GPCR 超家族的当前结构覆盖范围以及对原子分辨率下受体-配体相互作用的理解。然后,我们介绍了基于结构的虚拟筛选的一般工作流程和在化学文库中发现 GPCR 配体的策略。我们通过总结基于实验结构的虚拟筛选的前瞻性应用来评估该研究领域的最新技术。讨论了识别具有特定功效和选择性特征的化合物的策略,说明了分子对接方法的机会和局限性。我们的概述表明,基于结构的虚拟筛选可以发现新的线索,并且对于追求下一代 GPCR 药物至关重要。我们通过总结基于实验结构的虚拟筛选的前瞻性应用来评估该研究领域的最新技术。讨论了识别具有特定功效和选择性特征的化合物的策略,说明了分子对接方法的机会和局限性。我们的概述表明,基于结构的虚拟筛选可以发现新的线索,并且对于追求下一代 GPCR 药物至关重要。我们通过总结基于实验结构的虚拟筛选的前瞻性应用来评估该研究领域的最新技术。讨论了识别具有特定功效和选择性特征的化合物的策略,说明了分子对接方法的机会和局限性。我们的概述表明,基于结构的虚拟筛选可以发现新的线索,并且对于追求下一代 GPCR 药物至关重要。










































 京公网安备 11010802027423号
京公网安备 11010802027423号