当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-principles study of closo-dodecaborates M2B12H12 (M = Li, Na, K) as solid-state electrolyte materials
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-11-30 , DOI: 10.1039/d1cp03215a A Akrouchi 1 , H Benzidi 2 , A Al-Shami 1, 3 , A El Kenz 1 , A Benyoussef 4 , A El Kharbachi 5 , O Mounkachi 1, 6
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-11-30 , DOI: 10.1039/d1cp03215a A Akrouchi 1 , H Benzidi 2 , A Al-Shami 1, 3 , A El Kenz 1 , A Benyoussef 4 , A El Kharbachi 5 , O Mounkachi 1, 6
Affiliation
|
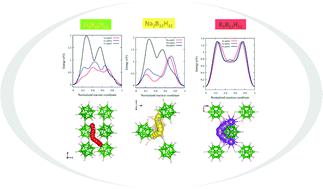
Closo-dodecaborates M2B12H12 are considered among the potential candidates for solid-state electrolyte materials due to their high ionic conductivities. It has been demonstrated that the reorientation of the icosahedral anion B12H122− plays a key role in high cation motion. However, this category of BnHn materials is still not well established with respect to their structural, thermodynamic and diffusion properties. In the present work, the electronic, vibrational and thermodynamic properties of M2B12H12 (M = Li, Na, K) structures are reported using first-principles calculations. The results of structural and electronic properties show that these structures have an insulator character with a large band gap of 5.75, 5.63 and 5.59 eV, respectively, for Li2B12H12, Na2B12H12 and K2B12H12. The thermodynamic stabilities of these systems are confirmed by their phonon calculation results. The primary quantities, such as heat capacity, vibrational entropy and volume variation at finite temperatures, are determined using the quasi-harmonic approximation in order to provide an input for the Gibbs free energy assessment. The calculated enthalpy of formation of the Li2B12H12 structure at 0 K and the proposed one at 300 K are found to be −127.31 and −740.44 kJ mol−1 per H2, respectively. The migration energy barrier of various cations in each system is calculated to be 0.7 (Li+), 1.16 (Na+) and 1.25 eV (K+), where the lowest energy barrier corresponds to the lithium ion migration in Li2B12H12. Additionally, the molecular dynamics simulation of M2B12H12 (M = Li, Na, K) structures demonstrated that these structures are stable above room temperature, except for the Li2B12H12 structure at 600 K, where the most stable is Na2B12H12. Finally, the temperature effect on icosahedral anion reorientation in each structure is elucidated as a function of temperature and cation type.
中文翻译:

近十二硼酸盐 M2B12H12 (M = Li, Na, K) 作为固态电解质材料的第一性原理研究
Closo- dodecaborates M 2 B 12 H 12由于其高离子电导率而被认为是固态电解质材料的潜在候选物。已经证明二十面体阴离子 B 12 H 12 2-的重新定向在高阳离子运动中起关键作用。然而,这类 B n H n材料在其结构、热力学和扩散特性方面仍未得到很好的确立。在目前的工作中,M 2 B 12 H 12的电子、振动和热力学性质(M = Li, Na, K) 结构是使用第一性原理计算报告的。结构和电子性质的结果表明,这些结构具有绝缘体特性,Li 2 B 12 H 12、Na 2 B 12 H 12和K 2 B 12 H 的带隙分别为5.75、5.63和5.59 eV。12. 这些系统的热力学稳定性由它们的声子计算结果证实。主要量,例如有限温度下的热容、振动熵和体积变化,是使用准谐波近似确定的,以便为吉布斯自由能评估提供输入。计算出的 Li 2 B 12 H 12结构在 0 K 和建议的在 300 K的形成焓分别为 -127.31 和 -740.44 kJ mol -1 per H 2。每个体系中各种阳离子的迁移能垒计算为0.7 (Li + )、1.16 (Na + )和1.25 eV (K +),其中最低能垒对应于 Li 2 B 12 H 12 中的锂离子迁移。此外,M 2 B 12 H 12 (M = Li, Na, K) 结构的分子动力学模拟表明,这些结构在室温以上是稳定的,除了 Li 2 B 12 H 12结构在 600 K 时,其中最稳定的是 Na 2 B 12 H 12。最后,阐明了温度对每个结构中二十面体阴离子重定向的影响,作为温度和阳离子类型的函数。
更新日期:2021-11-30
中文翻译:
近十二硼酸盐 M2B12H12 (M = Li, Na, K) 作为固态电解质材料的第一性原理研究
Closo- dodecaborates M 2 B 12 H 12由于其高离子电导率而被认为是固态电解质材料的潜在候选物。已经证明二十面体阴离子 B 12 H 12 2-的重新定向在高阳离子运动中起关键作用。然而,这类 B n H n材料在其结构、热力学和扩散特性方面仍未得到很好的确立。在目前的工作中,M 2 B 12 H 12的电子、振动和热力学性质(M = Li, Na, K) 结构是使用第一性原理计算报告的。结构和电子性质的结果表明,这些结构具有绝缘体特性,Li 2 B 12 H 12、Na 2 B 12 H 12和K 2 B 12 H 的带隙分别为5.75、5.63和5.59 eV。12. 这些系统的热力学稳定性由它们的声子计算结果证实。主要量,例如有限温度下的热容、振动熵和体积变化,是使用准谐波近似确定的,以便为吉布斯自由能评估提供输入。计算出的 Li 2 B 12 H 12结构在 0 K 和建议的在 300 K的形成焓分别为 -127.31 和 -740.44 kJ mol -1 per H 2。每个体系中各种阳离子的迁移能垒计算为0.7 (Li + )、1.16 (Na + )和1.25 eV (K +),其中最低能垒对应于 Li 2 B 12 H 12 中的锂离子迁移。此外,M 2 B 12 H 12 (M = Li, Na, K) 结构的分子动力学模拟表明,这些结构在室温以上是稳定的,除了 Li 2 B 12 H 12结构在 600 K 时,其中最稳定的是 Na 2 B 12 H 12。最后,阐明了温度对每个结构中二十面体阴离子重定向的影响,作为温度和阳离子类型的函数。










































 京公网安备 11010802027423号
京公网安备 11010802027423号