当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identifying the true origins of selectivity in chiral phosphoric acid catalyzed N-acyl-azetidine desymmetrizations
Chemical Science ( IF 7.6 ) Pub Date : 2021-11-23 , DOI: 10.1039/d1sc04969k Pier Alexandre Champagne 1
Chemical Science ( IF 7.6 ) Pub Date : 2021-11-23 , DOI: 10.1039/d1sc04969k Pier Alexandre Champagne 1
Affiliation
|
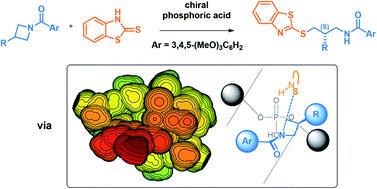
The first catalytic intermolecular desymmetrization of azetidines was reported by Sun and coworkers in 2015 using a BINOL-derived phosphoric acid catalyst (J. Am. Chem. Soc. 2015, 137, 5895–5898). To uncover the mechanism of the reaction and the origins of the high enantioselectivity, Density Functional Theory (DFT) calculations were performed at the B97D3/6-311+G(2d,2p)/SMD(toluene)//B97D3/6-31G(d,p)/CPCM(toluene) level of theory. Comparison of four possible activation modes confirms that this reaction proceeds through the bifunctional activation of the azetidine nitrogen and the thione tautomer of the 2-mercaptobenzothiazole nucleophile. Upon thorough conformational sampling of the enantiodetermining transition structures (TSs), a free energy difference of 2.0 kcal mol−1 is obtained, accurately reproducing the experimentally measured 88% e.e. at 80 °C. This energy difference is due to both decreased distortion and increased non-covalent interactions in the pro-(S) TS. To uncover the true origins of selectivity, the TSs optimized with the full catalyst were compared to those optimized with a model catalyst through steric maps. It is found that the arrangements displayed by the substrates are controlled by strict primary orbital interaction requirements at the transition complex, and their ability to fit into the catalyst pocket drives the selectivity. A general model of selectivity for phosphoric acid-catalyzed azetidine desymmetrizations is proposed, which is based on the preference of the nucleophile and benzoyl group to occupy empty quadrants of the chiral catalyst pocket.
中文翻译:

确定手性磷酸催化 N-酰基氮杂环丁烷去对称化选择性的真正起源
Sun 及其同事于 2015 年使用 BINOL 衍生的磷酸催化剂首次报道了氮杂环丁烷的催化分子间去对称化( J. Am. Chem. Soc. 2015, 137 , 5895–5898)。为了揭示反应机理和高对映选择性的起源,在 B97D3/6-311+G(2d,2p)/SMD(toluene)//B97D3/6-31G 上进行了密度泛函理论 (DFT) 计算(d,p)/CPCM(甲苯)理论水平。四种可能的活化模式的比较证实,该反应是通过氮杂环丁烷氮和 2-巯基苯并噻唑亲核试剂的硫酮互变异构体的双功能活化进行的。对映体决定过渡结构 (TS) 进行彻底的构象采样后,获得了 2.0 kcal mol -1的自由能差,准确地再现了 80 °C 下实验测量的 88% ee。这种能量差异是由于 pro-( S ) TS 中畸变的减少和非共价相互作用的增加所致。为了揭示选择性的真正来源,通过空间图将使用全催化剂优化的 TS 与使用模型催化剂优化的 TS 进行了比较。研究发现,底物显示的排列受到过渡配合物处严格的主轨道相互作用要求的控制,并且它们适合催化剂袋的能力驱动了选择性。提出了磷酸催化氮杂环丁烷不对称选择性的通用模型,该模型基于亲核试剂和苯甲酰基优先占据手性催化剂袋的空象限。
更新日期:2021-11-23
中文翻译:
确定手性磷酸催化 N-酰基氮杂环丁烷去对称化选择性的真正起源
Sun 及其同事于 2015 年使用 BINOL 衍生的磷酸催化剂首次报道了氮杂环丁烷的催化分子间去对称化( J. Am. Chem. Soc. 2015, 137 , 5895–5898)。为了揭示反应机理和高对映选择性的起源,在 B97D3/6-311+G(2d,2p)/SMD(toluene)//B97D3/6-31G 上进行了密度泛函理论 (DFT) 计算(d,p)/CPCM(甲苯)理论水平。四种可能的活化模式的比较证实,该反应是通过氮杂环丁烷氮和 2-巯基苯并噻唑亲核试剂的硫酮互变异构体的双功能活化进行的。对映体决定过渡结构 (TS) 进行彻底的构象采样后,获得了 2.0 kcal mol -1的自由能差,准确地再现了 80 °C 下实验测量的 88% ee。这种能量差异是由于 pro-( S ) TS 中畸变的减少和非共价相互作用的增加所致。为了揭示选择性的真正来源,通过空间图将使用全催化剂优化的 TS 与使用模型催化剂优化的 TS 进行了比较。研究发现,底物显示的排列受到过渡配合物处严格的主轨道相互作用要求的控制,并且它们适合催化剂袋的能力驱动了选择性。提出了磷酸催化氮杂环丁烷不对称选择性的通用模型,该模型基于亲核试剂和苯甲酰基优先占据手性催化剂袋的空象限。










































 京公网安备 11010802027423号
京公网安备 11010802027423号