当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Conversion of cyclic xylose into xylitol on Ru, Pt, Pd, Ni, and Rh catalysts: a density functional theory study
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2021-11-17 , DOI: 10.1039/d1cp04660h Shedrack G Akpe 1 , Sun Hee Choi 2 , Hyung Chul Ham 1, 3
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2021-11-17 , DOI: 10.1039/d1cp04660h Shedrack G Akpe 1 , Sun Hee Choi 2 , Hyung Chul Ham 1, 3
Affiliation
|
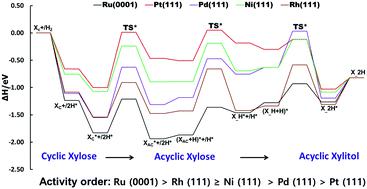
There is currently no theoretical study on the hydrogenation of xylose to xylitol on a catalyst's surface, limiting proper understanding of the reaction mechanisms and the design of effective catalysts. In this study, DFT techniques were used for the first time to investigate the mechanisms of xylose to xylitol conversion on five notable transition metal (TM) surfaces: Ru(0001), Pt(111), Pd(111), Rh(111), and Ni(111). Two transition state (TS) paths were investigated: TS Path A and TS Path B. The TS Path B, which was further subdivided into TS Path B1 and B2, was proposed to be the minimum energy path (MEP) for the reaction process. According to our computational results, the MEP for this reaction begins with the structural rearrangement of cyclic xylose into its acyclic form prior to step-wise hydrogenation. The rate-determining step (RDS) on Ru(0001), Pt(111), Pd(111), and Ni(111) was discovered to be the ring-opening process via C–O bond scission of cyclic xylose. On Rh(111), however, the RDS was found to be the first hydrogenation stage, leading to the hydrogenation intermediate. Furthermore, based on the RDS barrier, our results revealed that the activities of the tested TM surfaces follow the trend: Ru(0001) > Rh(111) ≥ Ni(111) > Pd(111) > Pt(111). This result demonstrates the higher activity of Ru(0001) compared to other surfaces used for xylose hydrogenation. It correlates with experimental trends in relation to Ru(0001) superiority and provides the basis for understanding the theoretical design of economical and more active catalysts for xylitol production.
中文翻译:

在 Ru、Pt、Pd、Ni 和 Rh 催化剂上将环状木糖转化为木糖醇:密度泛函理论研究
目前没有关于在催化剂表面木糖氢化成木糖醇的理论研究,限制了对反应机理的正确理解和有效催化剂的设计。在这项研究中,首次使用 DFT 技术研究了木糖在五种值得注意的过渡金属 (TM) 表面上转化为木糖醇的机制:Ru(0001)、Pt(111)、Pd(111)、Rh(111) ,和镍(111)。研究了两个过渡态 (TS) 路径:TS 路径 A 和 TS 路径 B。TS 路径 B 进一步细分为 TS 路径 B1 和 B2,被提议作为反应过程的最小能量路径 (MEP)。根据我们的计算结果,该反应的 MEP 始于在逐步氢化之前将环状木糖结构重排成其无环形式。通过环状木糖的 C-O 键断裂。然而,在 Rh(111) 上,发现 RDS 是第一个氢化阶段,导致氢化中间体。此外,基于 RDS 势垒,我们的结果显示测试的 TM 表面的活性遵循以下趋势:Ru(0001) > Rh(111) ≥ Ni(111) > Pd(111) > Pt(111)。该结果表明,与用于木糖氢化的其他表面相比,Ru(0001) 的活性更高。它与与 Ru(0001) 优势相关的实验趋势相关,并为理解用于木糖醇生产的经济且活性更高的催化剂的理论设计提供了基础。
更新日期:2021-11-23
中文翻译:
在 Ru、Pt、Pd、Ni 和 Rh 催化剂上将环状木糖转化为木糖醇:密度泛函理论研究
目前没有关于在催化剂表面木糖氢化成木糖醇的理论研究,限制了对反应机理的正确理解和有效催化剂的设计。在这项研究中,首次使用 DFT 技术研究了木糖在五种值得注意的过渡金属 (TM) 表面上转化为木糖醇的机制:Ru(0001)、Pt(111)、Pd(111)、Rh(111) ,和镍(111)。研究了两个过渡态 (TS) 路径:TS 路径 A 和 TS 路径 B。TS 路径 B 进一步细分为 TS 路径 B1 和 B2,被提议作为反应过程的最小能量路径 (MEP)。根据我们的计算结果,该反应的 MEP 始于在逐步氢化之前将环状木糖结构重排成其无环形式。通过环状木糖的 C-O 键断裂。然而,在 Rh(111) 上,发现 RDS 是第一个氢化阶段,导致氢化中间体。此外,基于 RDS 势垒,我们的结果显示测试的 TM 表面的活性遵循以下趋势:Ru(0001) > Rh(111) ≥ Ni(111) > Pd(111) > Pt(111)。该结果表明,与用于木糖氢化的其他表面相比,Ru(0001) 的活性更高。它与与 Ru(0001) 优势相关的实验趋势相关,并为理解用于木糖醇生产的经济且活性更高的催化剂的理论设计提供了基础。


























 京公网安备 11010802027423号
京公网安备 11010802027423号