当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular excited state calculations with adaptive wavefunctions on a quantum eigensolver emulation: reducing circuit depth and separating spin states
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-11-12 , DOI: 10.1039/d1cp02227j Hans Hon Sang Chan 1 , Nathan Fitzpatrick 2 , Javier Segarra-Martí 3 , Michael J Bearpark 4 , David P Tew 5
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-11-12 , DOI: 10.1039/d1cp02227j Hans Hon Sang Chan 1 , Nathan Fitzpatrick 2 , Javier Segarra-Martí 3 , Michael J Bearpark 4 , David P Tew 5
Affiliation
|
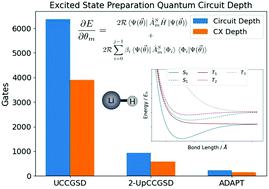
Ab initio electronic excited state calculations are necessary for the quantitative study of photochemical reactions, but their accurate computation on classical computers is plagued by prohibitive resource scaling. The Variational Quantum Deflation (VQD) is an extension of the quantum-classical Variational Quantum Eigensolver (VQE) algorithm for calculating electronic excited state energies, and has the potential to address some of these scaling challenges using quantum computers. However, quantum computers available in the near term can only support a limited number of quantum circuit operations, so reducing the quantum computational cost in VQD methods is critical to their realisation. In this work, we investigate the use of adaptive quantum circuit growth (ADAPT-VQE) in excited state VQD calculations, a strategy that has been successful previously in reducing the resources required for ground state energy VQE calculations. We also invoke spin restrictions to separate the recovery of eigenstates with different spin symmetry to reduce the number of calculations and accumulation of errors in computing excited states. We created a quantum eigensolver emulation package - Quantum Eigensolver Building on Achievements of Both quantum computing and quantum chemistry (QEBAB) – for testing the proposed adaptive procedure against two existing VQD methods that use fixed-length quantum circuits: UCCGSD-VQD and k-UpCCGSD-VQD. For a lithium hydride test case we found that the spin-restricted adaptive growth variant of VQD uses the most compact circuits out of the tested methods by far, consistently recovers adequate electron correlation energy for different nuclear geometries and eigenstates while isolating the singlet and triplet manifold. This work is a further step towards developing techniques which improve the efficiency of hybrid quantum algorithms for excited state quantum chemistry, opening up the possibility of exploiting real quantum computers for electronic excited state calculations sooner than previously anticipated.
中文翻译:

在量子本征求解器仿真中使用自适应波函数进行分子激发态计算:减少电路深度和分离自旋态
从头开始电子激发态计算对于光化学反应的定量研究是必要的,但它们在经典计算机上的准确计算受到令人望而却步的资源缩放的困扰。变分量子紧缩 (VQD) 是用于计算电子激发态能量的量子经典变分量子特征求解器 (VQE) 算法的扩展,并且有可能使用量子计算机解决其中的一些缩放挑战。然而,近期可用的量子计算机只能支持有限数量的量子电路操作,因此降低 VQD 方法中的量子计算成本对其实现至关重要。在这项工作中,我们研究了自适应量子电路增长 (ADAPT-VQE) 在激发态 VQD 计算中的使用,以前在减少基态能量 VQE 计算所需资源方面取得成功的策略。我们还调用自旋限制来分离具有不同自旋对称性的本征态的恢复,以减少计算次数和计算激发态的错误累积。我们创建了一个量子特征求解器仿真包——基于量子计算和量子化学成就 (QEBAB) 的量子特征求解器——用于针对使用固定长度量子电路的两种现有 VQD 方法测试提议的自适应程序:UCCGSD-VQD 和 我们还调用自旋限制来分离具有不同自旋对称性的本征态的恢复,以减少计算次数和计算激发态的错误累积。我们创建了一个量子特征求解器仿真包——基于量子计算和量子化学成就 (QEBAB) 的量子特征求解器——用于针对使用固定长度量子电路的两种现有 VQD 方法测试提议的自适应程序:UCCGSD-VQD 和 我们还调用自旋限制来分离具有不同自旋对称性的本征态的恢复,以减少计算次数和计算激发态的错误累积。我们创建了一个量子特征求解器仿真包——基于量子计算和量子化学成就 (QEBAB) 的量子特征求解器——用于针对使用固定长度量子电路的两种现有 VQD 方法测试提议的自适应程序:UCCGSD-VQD 和k -UpCCGSD-VQD。对于氢化锂测试案例,我们发现 VQD 的自旋受限自适应增长变体使用迄今为止测试方法中最紧凑的电路,在隔离单线态和三线态流形的同时,始终为不同的核几何形状和本征态恢复足够的电子相关能. 这项工作是朝着开发提高激发态量子化学混合量子算法效率的技术迈出的又一步,开辟了利用真正的量子计算机进行电子激发态计算的可能性,比以前预期的要早。
更新日期:2021-11-22
中文翻译:
在量子本征求解器仿真中使用自适应波函数进行分子激发态计算:减少电路深度和分离自旋态
从头开始电子激发态计算对于光化学反应的定量研究是必要的,但它们在经典计算机上的准确计算受到令人望而却步的资源缩放的困扰。变分量子紧缩 (VQD) 是用于计算电子激发态能量的量子经典变分量子特征求解器 (VQE) 算法的扩展,并且有可能使用量子计算机解决其中的一些缩放挑战。然而,近期可用的量子计算机只能支持有限数量的量子电路操作,因此降低 VQD 方法中的量子计算成本对其实现至关重要。在这项工作中,我们研究了自适应量子电路增长 (ADAPT-VQE) 在激发态 VQD 计算中的使用,以前在减少基态能量 VQE 计算所需资源方面取得成功的策略。我们还调用自旋限制来分离具有不同自旋对称性的本征态的恢复,以减少计算次数和计算激发态的错误累积。我们创建了一个量子特征求解器仿真包——基于量子计算和量子化学成就 (QEBAB) 的量子特征求解器——用于针对使用固定长度量子电路的两种现有 VQD 方法测试提议的自适应程序:UCCGSD-VQD 和 我们还调用自旋限制来分离具有不同自旋对称性的本征态的恢复,以减少计算次数和计算激发态的错误累积。我们创建了一个量子特征求解器仿真包——基于量子计算和量子化学成就 (QEBAB) 的量子特征求解器——用于针对使用固定长度量子电路的两种现有 VQD 方法测试提议的自适应程序:UCCGSD-VQD 和 我们还调用自旋限制来分离具有不同自旋对称性的本征态的恢复,以减少计算次数和计算激发态的错误累积。我们创建了一个量子特征求解器仿真包——基于量子计算和量子化学成就 (QEBAB) 的量子特征求解器——用于针对使用固定长度量子电路的两种现有 VQD 方法测试提议的自适应程序:UCCGSD-VQD 和k -UpCCGSD-VQD。对于氢化锂测试案例,我们发现 VQD 的自旋受限自适应增长变体使用迄今为止测试方法中最紧凑的电路,在隔离单线态和三线态流形的同时,始终为不同的核几何形状和本征态恢复足够的电子相关能. 这项工作是朝着开发提高激发态量子化学混合量子算法效率的技术迈出的又一步,开辟了利用真正的量子计算机进行电子激发态计算的可能性,比以前预期的要早。










































 京公网安备 11010802027423号
京公网安备 11010802027423号