当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Quantum Dynamics: A Quantum Computing Perspective
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2021-11-17 , DOI: 10.1021/acs.accounts.1c00514 Pauline J Ollitrault 1 , Alexander Miessen 1 , Ivano Tavernelli 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2021-11-17 , DOI: 10.1021/acs.accounts.1c00514 Pauline J Ollitrault 1 , Alexander Miessen 1 , Ivano Tavernelli 1
Affiliation
|
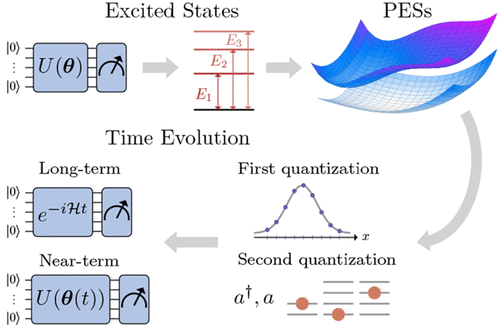
Simulating molecular dynamics (MD) within a comprehensive quantum framework has been a long-standing challenge in computational chemistry. An exponential scaling of computational cost renders solving the time dependent Schrödinger equation (TDSE) of a molecular Hamiltonian, including both electronic and nuclear degrees of freedom (DOFs), as well as their couplings, infeasible for more than a few DOFs. In the Born–Oppenheimer (BO), or adiabatic, picture, electronic and nuclear parts of the wave function are decoupled and treated separately. Within this framework, the nuclear wave function evolves along potential energy surfaces (PESs) computed as solutions to the electronic Schrödinger equation parametrized in the nuclear DOFs. This approximation, together with increasingly elaborate numerical approaches to solve the nuclear time dependent Schrödinger equation (TDSE), enabled the treatment of up to a few dozens of degrees of freedom (DOFs). However, for particular applications, such as photochemistry, the BO approximation breaks down. In this regime of non-adiabatic dynamics, solving the full molecular problem including electron–nuclear couplings becomes essential, further increasing the complexity of the numerical solution. Although valuable methods such as multiconfigurational time-dependent Hartree (MCTDH) have been proposed for the solution of the coupled electron–nuclear dynamics, they remain hampered by an exponential scaling in the number of nuclear DOFs and by the difficulty of finding universal variational forms.
中文翻译:

分子量子动力学:量子计算视角
在综合量子框架内模拟分子动力学 (MD) 一直是计算化学领域的长期挑战。计算成本的指数缩放使得求解分子哈密顿量的时间相关薛定谔方程 (TDSE),包括电子和核自由度 (DOF) 以及它们的耦合,对于多个 DOF 而言是不可行的。在波恩-奥本海默 (BO) 或绝热图像中,波函数的电子部分和核部分被解耦并分别处理。在此框架内,核波函数沿势能面 (PES) 演化,计算为在核自由度中参数化的电子薛定谔方程的解。这个近似,再加上越来越精细的数值方法来求解核时间相关薛定谔方程 (TDSE),能够处理多达几十个自由度 (DOF)。但是,对于特定应用,例如光化学,BO 近似会失效。在这种非绝热动力学领域,解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。能够处理多达几十个自由度 (DOF)。但是,对于特定应用,例如光化学,BO 近似会失效。在这种非绝热动力学领域,解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。能够处理多达几十个自由度 (DOF)。但是,对于特定应用,例如光化学,BO 近似会失效。在这种非绝热动力学领域,解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。
更新日期:2021-12-07
中文翻译:
分子量子动力学:量子计算视角
在综合量子框架内模拟分子动力学 (MD) 一直是计算化学领域的长期挑战。计算成本的指数缩放使得求解分子哈密顿量的时间相关薛定谔方程 (TDSE),包括电子和核自由度 (DOF) 以及它们的耦合,对于多个 DOF 而言是不可行的。在波恩-奥本海默 (BO) 或绝热图像中,波函数的电子部分和核部分被解耦并分别处理。在此框架内,核波函数沿势能面 (PES) 演化,计算为在核自由度中参数化的电子薛定谔方程的解。这个近似,再加上越来越精细的数值方法来求解核时间相关薛定谔方程 (TDSE),能够处理多达几十个自由度 (DOF)。但是,对于特定应用,例如光化学,BO 近似会失效。在这种非绝热动力学领域,解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。能够处理多达几十个自由度 (DOF)。但是,对于特定应用,例如光化学,BO 近似会失效。在这种非绝热动力学领域,解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。能够处理多达几十个自由度 (DOF)。但是,对于特定应用,例如光化学,BO 近似会失效。在这种非绝热动力学领域,解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。解决包括电子-核耦合在内的全分子问题变得至关重要,这进一步增加了数值解的复杂性。尽管已经提出了诸如多构型时间相关 Hartree (MCTDH) 等有价值的方法来解决耦合电子-核动力学问题,但它们仍然受到核自由度数量的指数缩放和难以找到通用变分形式的阻碍。










































 京公网安备 11010802027423号
京公网安备 11010802027423号