当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanisms of the Cu(I)-Catalyzed Intermolecular Photocycloaddition Reaction Revealed by Optical and X-ray Transient Absorption Spectroscopies
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2021-11-09 , DOI: 10.1021/jacs.1c07282 Gethmini K Jayasekara 1 , Cali Antolini 1 , Melissa A Smith 1 , Danielle J Jacoby 1 , Jacqueline Escolastico 2 , Nathan Girard 2 , Benjamin T Young 2 , Dugan Hayes 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2021-11-09 , DOI: 10.1021/jacs.1c07282 Gethmini K Jayasekara 1 , Cali Antolini 1 , Melissa A Smith 1 , Danielle J Jacoby 1 , Jacqueline Escolastico 2 , Nathan Girard 2 , Benjamin T Young 2 , Dugan Hayes 1
Affiliation
|
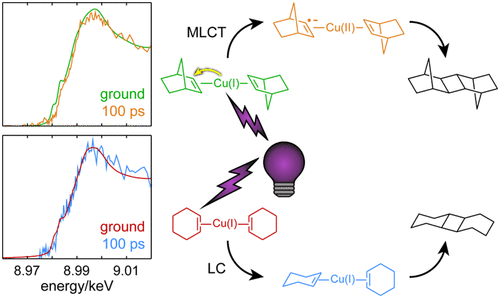
The [2 + 2] photocycloaddition provides a simple, single-step route to cyclobutane moieties that would otherwise be disfavored or impossible due to ring strain and/or steric interactions. We have used a combination of optical and X-ray transient absorption spectroscopies to elucidate the mechanism of the Cu(I)-catalyzed intermolecular photocycloaddition reaction using norbornene and cyclohexene as model substrates. We find that for norbornene the reaction proceeds through an initial metal-to-ligand charge transfer (MLCT) state that persists for 18 ns before the metal returns to the monovalent oxidation state. The Cu K-edge spectrum continues to evolve until ∼5 μs and then remains unchanged for the 50 μs duration of the measurement, reflecting product formation and ligand dissociation. We hypothesize that the MLCT transition and reverse electron transfer serve to sensitize the triplet excited state of one of the norbornene ligands, which then dimerizes with the other to give the product. For the case of cyclohexene, however, we do not observe a charge transfer state following photoexcitation and instead find evidence for an increase in the metal–ligand bond strength that persists for several ns before product formation occurs. This is consistent with a mechanism in which ligand photoisomerization is the initial step, which was first proposed by Salomon and Kochi in 1974 to explain the stereoselectivity of the reaction. Our investigation reveals how this photocatalytic reaction may be directed along strikingly disparate trajectories by only very minor changes to the structure of the substrate.
中文翻译:

光学和 X 射线瞬态吸收光谱揭示 Cu(I) 催化的分子间光环加成反应机理
[2 + 2] 光环加成为环丁烷部分提供了一种简单的单步路线,否则由于环应变和/或空间相互作用,环丁烷部分将不受欢迎或不可能。我们结合使用光学和 X 射线瞬态吸收光谱来阐明使用降冰片烯和环己烯作为模型底物的 Cu(I) 催化的分子间光环加成反应的机理。我们发现,对于降冰片烯,反应通过初始金属-配体电荷转移 (MLCT) 状态进行,该状态持续 18 ns,然后金属返回单价氧化态。Cu K-edge 光谱继续演化直到 5 μs,然后在 50 μs 测量期间保持不变,反映产物形成和配体解离。我们假设 MLCT 跃迁和反向电子转移有助于使降冰片烯配体之一的三重激发态敏感,然后与另一个二聚体形成产物。然而,对于环己烯,我们没有观察到光激发后的电荷转移状态,而是发现了金属-配体键强度增加的证据,该强度在产物形成之前持续了数 ns。这与配体光异构化是初始步骤的机制是一致的,该机制由 Salomon 和 Kochi 在 1974 年首次提出以解释反应的立体选择性。我们的研究揭示了这种光催化反应是如何通过对底物结构进行非常微小的改变而沿着截然不同的轨迹进行的。
更新日期:2021-11-24
中文翻译:
光学和 X 射线瞬态吸收光谱揭示 Cu(I) 催化的分子间光环加成反应机理
[2 + 2] 光环加成为环丁烷部分提供了一种简单的单步路线,否则由于环应变和/或空间相互作用,环丁烷部分将不受欢迎或不可能。我们结合使用光学和 X 射线瞬态吸收光谱来阐明使用降冰片烯和环己烯作为模型底物的 Cu(I) 催化的分子间光环加成反应的机理。我们发现,对于降冰片烯,反应通过初始金属-配体电荷转移 (MLCT) 状态进行,该状态持续 18 ns,然后金属返回单价氧化态。Cu K-edge 光谱继续演化直到 5 μs,然后在 50 μs 测量期间保持不变,反映产物形成和配体解离。我们假设 MLCT 跃迁和反向电子转移有助于使降冰片烯配体之一的三重激发态敏感,然后与另一个二聚体形成产物。然而,对于环己烯,我们没有观察到光激发后的电荷转移状态,而是发现了金属-配体键强度增加的证据,该强度在产物形成之前持续了数 ns。这与配体光异构化是初始步骤的机制是一致的,该机制由 Salomon 和 Kochi 在 1974 年首次提出以解释反应的立体选择性。我们的研究揭示了这种光催化反应是如何通过对底物结构进行非常微小的改变而沿着截然不同的轨迹进行的。









































 京公网安备 11010802027423号
京公网安备 11010802027423号