当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, synthesis, structural analysis and quantum chemical insight into the molecular structure of coumarin derivatives
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2021-11-03 , DOI: 10.1039/d1me00113b Chethan Burudeghatta Sundaramurthy 1 , Chethan Prathap Kesthur Nataraju 2 , Lokanath Neratur Krishnappagowda 1
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2021-11-03 , DOI: 10.1039/d1me00113b Chethan Burudeghatta Sundaramurthy 1 , Chethan Prathap Kesthur Nataraju 2 , Lokanath Neratur Krishnappagowda 1
Affiliation
|
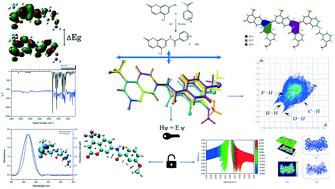
The aim of the current research article is to present the supramolecular features in the crystal structure of seven novel coumarin derivatives, their structural modification due to the addition of various functional groups and the results of spectroscopic investigations as well as theoretical calculations. The compounds were characterized by NMR, FTIR, UV-vis, mass spectroscopy, and single crystal X-ray diffraction studies. The structural analysis revealed that the coumarin moiety exhibits planarity in all the compounds with average dihedral angles of 1.71(2)° between the pyran and fused benzene rings and 9.40(9)° between the benzopyran and terminal benzene rings. Various supramolecular architectures formed due to the intermolecular interactions significantly contribute to the crystal packing of the molecules. The intermolecular interactions and their contributions were quantified using 2D fingerprint plots. The structural aspects of the compounds were examined based on their optimized geometry, intramolecular hydrogen bonding and chemical reactivity using density functional theory. Theoretical FTIR calculations were performed using VEDA4 software and the TD-DFT method was used to study the electronic transitions of the compounds. The theoretical FTIR and UV data were compared with the experimental data. Natural bond orbital (NBO) analysis revealed the presence and the importance of non-covalent and hyperconjugative interactions for the stability of the molecules in their solid state. The charge distribution and nucleophilic and electrophilic regions of the molecules were identified by molecular electrostatic potential (MEP) mapping. A quantum theory of atoms in molecules (QTAIM) study was carried out to investigate the nature and strength of the hydrogen bonding interactions.
中文翻译:

香豆素衍生物分子结构的设计、合成、结构分析和量子化学洞察
当前研究文章的目的是介绍七种新型香豆素衍生物的晶体结构中的超分子特征、由于添加各种官能团而导致的结构变化以及光谱研究和理论计算的结果。通过NMR、FTIR、UV-vis、质谱和单晶X射线衍射研究表征化合物。结构分析表明,香豆素部分在所有化合物中表现出平面性,吡喃和稠合苯环之间的平均二面角为 1.71(2)°,苯并吡喃和末端苯环之间的平均二面角为 9.40(9)°。由于分子间相互作用而形成的各种超分子结构显着促进了分子的晶体堆积。分子间相互作用及其贡献使用二维指纹图进行量化。使用密度泛函理论,基于化合物的优化几何形状、分子内氢键和化学反应性来检查化合物的结构方面。使用 VEDA4 软件进行理论 FTIR 计算,并使用 TD-DFT 方法研究化合物的电子跃迁。将理论 FTIR 和 UV 数据与实验数据进行比较。自然键轨道 (NBO) 分析揭示了非共价和超共轭相互作用对固态分子稳定性的存在和重要性。通过分子静电势 (MEP) 映射确定分子的电荷分布和亲核和亲电区域。
更新日期:2021-11-24
中文翻译:
香豆素衍生物分子结构的设计、合成、结构分析和量子化学洞察
当前研究文章的目的是介绍七种新型香豆素衍生物的晶体结构中的超分子特征、由于添加各种官能团而导致的结构变化以及光谱研究和理论计算的结果。通过NMR、FTIR、UV-vis、质谱和单晶X射线衍射研究表征化合物。结构分析表明,香豆素部分在所有化合物中表现出平面性,吡喃和稠合苯环之间的平均二面角为 1.71(2)°,苯并吡喃和末端苯环之间的平均二面角为 9.40(9)°。由于分子间相互作用而形成的各种超分子结构显着促进了分子的晶体堆积。分子间相互作用及其贡献使用二维指纹图进行量化。使用密度泛函理论,基于化合物的优化几何形状、分子内氢键和化学反应性来检查化合物的结构方面。使用 VEDA4 软件进行理论 FTIR 计算,并使用 TD-DFT 方法研究化合物的电子跃迁。将理论 FTIR 和 UV 数据与实验数据进行比较。自然键轨道 (NBO) 分析揭示了非共价和超共轭相互作用对固态分子稳定性的存在和重要性。通过分子静电势 (MEP) 映射确定分子的电荷分布和亲核和亲电区域。









































 京公网安备 11010802027423号
京公网安备 11010802027423号