当前位置:
X-MOL 学术
›
Comput. Struct. Biotechnol. J.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A k-mer based approach for classifying viruses without taxonomy identifies viral associations in human autism and plant microbiomes
Computational and Structural Biotechnology Journal ( IF 4.4 ) Pub Date : 2021-10-25 , DOI: 10.1016/j.csbj.2021.10.029 Benjamin J Garcia 1, 2, 3 , Ramanuja Simha 1 , Michael Garvin 1 , Anna Furches 1, 4 , Piet Jones 1, 4 , Joao G F M Gazolla 1 , P Doug Hyatt 1 , Christopher W Schadt 1 , Dale Pelletier 1 , Daniel Jacobson 1, 4
Computational and Structural Biotechnology Journal ( IF 4.4 ) Pub Date : 2021-10-25 , DOI: 10.1016/j.csbj.2021.10.029 Benjamin J Garcia 1, 2, 3 , Ramanuja Simha 1 , Michael Garvin 1 , Anna Furches 1, 4 , Piet Jones 1, 4 , Joao G F M Gazolla 1 , P Doug Hyatt 1 , Christopher W Schadt 1 , Dale Pelletier 1 , Daniel Jacobson 1, 4
Affiliation
|
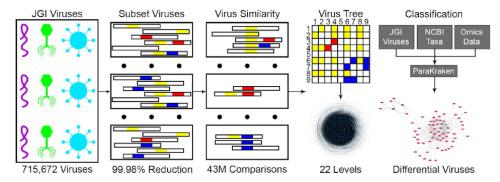
Viruses are an underrepresented taxa in the study and identification of microbiome constituents; however, they play an essential role in health, microbiome regulation, and transfer of genetic material. Only a few thousand viruses have been isolated, sequenced, and assigned a taxonomy, which limits the ability to identify and quantify viruses in the microbiome. Additionally, the vast diversity of viruses represents a challenge for classification, not only in constructing a viral taxonomy, but also in identifying similarities between a virus’ genotype and its phenotype. However, the diversity of viral sequences can be leveraged to classify their sequences in metagenomic and metatranscriptomic samples, even if they do not have a taxonomy. To identify and quantify viruses in transcriptomic and genomic samples, we developed a dynamic programming algorithm for creating a classification tree out of 715,672 metagenome viruses. To create the classification tree, we clustered proportional similarity scores generated from the k-mer profiles of each of the metagenome viruses to create a database of metagenomic viruses. The resulting Kraken2 database of the metagenomic viruses can be found here: and is compatible with Kraken2. We then integrated the viral classification database with databases created with genomes from NCBI for use with ParaKraken (a parallelized version of Kraken provided in Supplemental Zip 1), a metagenomic/transcriptomic classifier. To illustrate the breadth of our utility for classifying metagenome viruses, we analyzed data from a plant metagenome study identifying genotypic and compartment specific differences between two genotypes in three different compartments. We also identified a significant increase in abundance of eight viral sequences in post mortem brains in a human metatranscriptome study comparing Autism Spectrum Disorder patients and controls. We also show the potential accuracy for classifying viruses by utilizing both the JGI and NCBI viral databases to identify the uniqueness of viral sequences. Finally, we validate the accuracy of viral classification with NCBI databases containing viruses with taxonomy to identify pathogenic viruses in known COVID-19 and cassava brown streak virus infection samples. Our method represents the compulsory first step in better understanding the role of viruses in the microbiome by allowing for a more complete identification of sequences without taxonomy. Better classification of viruses will improve identifying associations between viruses and their hosts as well as viruses and other microbiome members. Despite the lack of taxonomy, this database of metagenomic viruses can be used with any tool that utilizes a taxonomy, such as Kraken, for accurate classification of viruses.
中文翻译:

基于 k-mer 的病毒分类方法无需分类学即可识别人类自闭症和植物微生物组中的病毒关联
在微生物组成分的研究和鉴定中,病毒是代表性不足的类群;然而,它们在健康、微生物组调节和遗传物质转移方面发挥着重要作用。只有几千种病毒被分离、测序并分配了分类法,这限制了识别和量化微生物组中病毒的能力。此外,病毒的巨大多样性对分类提出了挑战,不仅在构建病毒分类学方面,而且在识别病毒基因型和表型之间的相似性方面。然而,可以利用病毒序列的多样性对宏基因组和宏转录组样本中的序列进行分类,即使它们没有分类法。为了识别和量化转录组和基因组样本中的病毒,我们开发了一种动态编程算法,用于从 715,672 种宏基因组病毒中创建分类树。为了创建分类树,我们对从每个宏基因组病毒的 k 聚体概况生成的比例相似性分数进行了聚类,以创建宏基因组病毒的数据库。生成的宏基因组病毒 Kraken2 数据库可以在此处找到:并且与 Kraken2 兼容。然后,我们将病毒分类数据库与使用 NCBI 基因组创建的数据库集成,以便与 ParaKraken(补充 Zip 1 中提供的 Kraken 的并行版本)(一种宏基因组/转录组分类器)一起使用。为了说明我们对宏基因组病毒进行分类的实用性的广度,我们分析了植物宏基因组研究的数据,识别了三个不同区室中两种基因型之间的基因型和区室特异性差异。 在一项比较自闭症谱系障碍患者和对照组的人类宏转录组研究中,我们还发现死后大脑中八种病毒序列的丰度显着增加。我们还通过利用 JGI 和 NCBI 病毒数据库来识别病毒序列的独特性,展示了病毒分类的潜在准确性。最后,我们使用包含病毒分类学的 NCBI 数据库验证了病毒分类的准确性,以识别已知的 COVID-19 和木薯褐条病毒感染样本中的致病病毒。我们的方法通过允许在不分类的情况下更完整地识别序列,代表了更好地理解病毒在微生物组中的作用的强制性第一步。更好的病毒分类将有助于识别病毒与其宿主以及病毒与其他微生物组成员之间的关联。尽管缺乏分类学,这个宏基因组病毒数据库可以与任何利用分类学的工具(例如 Kraken)一起使用,以对病毒进行准确分类。
更新日期:2021-10-25
中文翻译:
基于 k-mer 的病毒分类方法无需分类学即可识别人类自闭症和植物微生物组中的病毒关联
在微生物组成分的研究和鉴定中,病毒是代表性不足的类群;然而,它们在健康、微生物组调节和遗传物质转移方面发挥着重要作用。只有几千种病毒被分离、测序并分配了分类法,这限制了识别和量化微生物组中病毒的能力。此外,病毒的巨大多样性对分类提出了挑战,不仅在构建病毒分类学方面,而且在识别病毒基因型和表型之间的相似性方面。然而,可以利用病毒序列的多样性对宏基因组和宏转录组样本中的序列进行分类,即使它们没有分类法。为了识别和量化转录组和基因组样本中的病毒,我们开发了一种动态编程算法,用于从 715,672 种宏基因组病毒中创建分类树。为了创建分类树,我们对从每个宏基因组病毒的 k 聚体概况生成的比例相似性分数进行了聚类,以创建宏基因组病毒的数据库。生成的宏基因组病毒 Kraken2 数据库可以在此处找到:并且与 Kraken2 兼容。然后,我们将病毒分类数据库与使用 NCBI 基因组创建的数据库集成,以便与 ParaKraken(补充 Zip 1 中提供的 Kraken 的并行版本)(一种宏基因组/转录组分类器)一起使用。为了说明我们对宏基因组病毒进行分类的实用性的广度,我们分析了植物宏基因组研究的数据,识别了三个不同区室中两种基因型之间的基因型和区室特异性差异。 在一项比较自闭症谱系障碍患者和对照组的人类宏转录组研究中,我们还发现死后大脑中八种病毒序列的丰度显着增加。我们还通过利用 JGI 和 NCBI 病毒数据库来识别病毒序列的独特性,展示了病毒分类的潜在准确性。最后,我们使用包含病毒分类学的 NCBI 数据库验证了病毒分类的准确性,以识别已知的 COVID-19 和木薯褐条病毒感染样本中的致病病毒。我们的方法通过允许在不分类的情况下更完整地识别序列,代表了更好地理解病毒在微生物组中的作用的强制性第一步。更好的病毒分类将有助于识别病毒与其宿主以及病毒与其他微生物组成员之间的关联。尽管缺乏分类学,这个宏基因组病毒数据库可以与任何利用分类学的工具(例如 Kraken)一起使用,以对病毒进行准确分类。










































 京公网安备 11010802027423号
京公网安备 11010802027423号