当前位置:
X-MOL 学术
›
J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design of a “Two-in-One” Mutant-Selective Epidermal Growth Factor Receptor Inhibitor That Spans the Orthosteric and Allosteric Sites
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2021-10-20 , DOI: 10.1021/acs.jmedchem.1c00848 Florian Wittlinger 1 , David E Heppner 2 , Ciric To 3 , Marcel Günther 1 , Bo Hee Shin 3 , Jaimin K Rana 2 , Anna M Schmoker 2 , Tyler S Beyett 2 , Lena M Berger 4, 5 , Benedict-Tilman Berger 4, 5 , Nicolas Bauer 4, 5 , James D Vasta 6 , Cesear R Corona 6 , Matthew B Robers 6 , Stefan Knapp 4, 5 , Pasi A Jänne 3, 7 , Michael J Eck 2 , Stefan A Laufer 1, 8, 9
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2021-10-20 , DOI: 10.1021/acs.jmedchem.1c00848 Florian Wittlinger 1 , David E Heppner 2 , Ciric To 3 , Marcel Günther 1 , Bo Hee Shin 3 , Jaimin K Rana 2 , Anna M Schmoker 2 , Tyler S Beyett 2 , Lena M Berger 4, 5 , Benedict-Tilman Berger 4, 5 , Nicolas Bauer 4, 5 , James D Vasta 6 , Cesear R Corona 6 , Matthew B Robers 6 , Stefan Knapp 4, 5 , Pasi A Jänne 3, 7 , Michael J Eck 2 , Stefan A Laufer 1, 8, 9
Affiliation
|
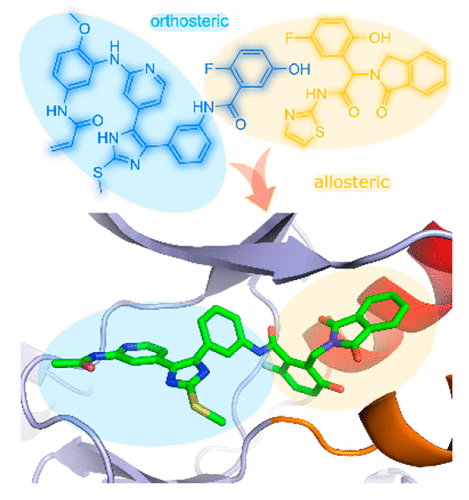
Inhibitors targeting the epidermal growth factor receptor (EGFR) are an effective therapy for patients with non-small cell lung cancer harboring drug-sensitive activating mutations in the EGFR kinase domain. Drug resistance due to treatment-acquired mutations has motivated the development of successive generations of inhibitors that bind in the ATP site. The third-generation agent osimertinib is now a first-line treatment for this disease. Recently, allosteric inhibitors have been developed to overcome drug-resistant mutations that confer a resistance to osimertinib. Here, we present the structure-guided design and synthesis of a mutant-selective lead compound, which consists of a pyridinyl imidazole-fused benzylisoindolinedione scaffold that simultaneously occupies the orthosteric and allosteric sites. The compound potently inhibits enzymatic activity in L858R/T790M/C797S mutant EGFR (4.9 nM), with a significantly lower activity for wild-type EGFR (47 nM). Additionally, this compound achieves modest cetuximab-independent and mutant-selective cellular efficacies on the L858R (1.2 μM) and L858R/T790M (4.4 μM) variants.
中文翻译:

跨越正构和变构位点的“二合一”突变选择性表皮生长因子受体抑制剂的设计
针对表皮生长因子受体 (EGFR) 的抑制剂是治疗 EGFR 激酶结构域中具有药物敏感性激活突变的非小细胞肺癌患者的有效疗法。由于治疗获得性突变而产生的耐药性推动了在 ATP 位点结合的连续几代抑制剂的开发。第三代药物奥希替尼现已成为该病的一线治疗药物。最近,已经开发出变构抑制剂来克服赋予奥希替尼耐药性的耐药突变。在这里,我们介绍了突变选择性先导化合物的结构指导设计和合成,该先导化合物由同时占据正构和变构位点的吡啶基咪唑稠合苄基异二氢吲哚二酮支架组成。该化合物有效抑制 L858R/T790M/C797S 突变体 EGFR 中的酶活性 (4.9 nM),对野生型 EGFR (47 nM) 的活性显着降低。此外,该化合物对 L858R (1.2 μM) 和 L858R/T790M (4.4 μM) 变体具有适度的非西妥昔单抗依赖性和突变体选择性细胞功效。
更新日期:2021-10-20
中文翻译:
跨越正构和变构位点的“二合一”突变选择性表皮生长因子受体抑制剂的设计
针对表皮生长因子受体 (EGFR) 的抑制剂是治疗 EGFR 激酶结构域中具有药物敏感性激活突变的非小细胞肺癌患者的有效疗法。由于治疗获得性突变而产生的耐药性推动了在 ATP 位点结合的连续几代抑制剂的开发。第三代药物奥希替尼现已成为该病的一线治疗药物。最近,已经开发出变构抑制剂来克服赋予奥希替尼耐药性的耐药突变。在这里,我们介绍了突变选择性先导化合物的结构指导设计和合成,该先导化合物由同时占据正构和变构位点的吡啶基咪唑稠合苄基异二氢吲哚二酮支架组成。该化合物有效抑制 L858R/T790M/C797S 突变体 EGFR 中的酶活性 (4.9 nM),对野生型 EGFR (47 nM) 的活性显着降低。此外,该化合物对 L858R (1.2 μM) 和 L858R/T790M (4.4 μM) 变体具有适度的非西妥昔单抗依赖性和突变体选择性细胞功效。










































 京公网安备 11010802027423号
京公网安备 11010802027423号