当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Drug Repurposing to Identify Nilotinib as a Potential SARS-CoV-2 Main Protease Inhibitor: Insights from a Computational and In Vitro Study
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-20 , DOI: 10.1021/acs.jcim.1c00524 Souvik Banerjee 1 , Shalini Yadav 2 , Sourav Banerjee 3 , Sayo O Fakayode 1 , Jyothi Parvathareddy 4 , Walter Reichard 5 , Surekha Surendranathan 4 , Foyez Mahmud 6 , Ryan Whatcott 1 , Joshua Thammathong 1 , Bernd Meibohm 7 , Duane D Miller 7 , Colleen B Jonsson 4, 5, 7 , Kshatresh Dutta Dubey 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-20 , DOI: 10.1021/acs.jcim.1c00524 Souvik Banerjee 1 , Shalini Yadav 2 , Sourav Banerjee 3 , Sayo O Fakayode 1 , Jyothi Parvathareddy 4 , Walter Reichard 5 , Surekha Surendranathan 4 , Foyez Mahmud 6 , Ryan Whatcott 1 , Joshua Thammathong 1 , Bernd Meibohm 7 , Duane D Miller 7 , Colleen B Jonsson 4, 5, 7 , Kshatresh Dutta Dubey 2
Affiliation
|
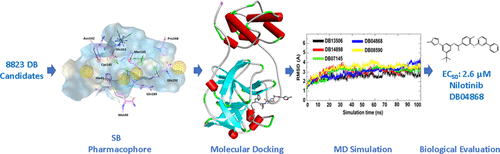
COVID-19, an acute viral pneumonia, has emerged as a devastating pandemic. Drug repurposing allows researchers to find different indications of FDA-approved or investigational drugs. In this current study, a sequence of pharmacophore and molecular modeling-based screening against COVID-19 Mpro (PDB: 6LU7) suggested a subset of drugs, from the Drug Bank database, which may have antiviral activity. A total of 44 out of 8823 of the most promising virtual hits from the Drug Bank were subjected to molecular dynamics simulation experiments to explore the strength of their interactions with the SARS-CoV-2 Mpro active site. MD findings point toward three drugs (DB04020, DB12411, and DB11779) with very low relative free energies for SARS-CoV-2 Mpro with interactions at His41 and Met49. MD simulations identified an additional interaction with Glu166, which enhanced the binding affinity significantly. Therefore, Glu166 could be an interesting target for structure-based drug design. Quantitative structural–activity relationship analysis was performed on the 44 most promising hits from molecular docking-based virtual screening. Partial least square regression accurately predicted the values of independent drug candidates’ binding energy with impressively high accuracy. Finally, the EC50 and CC50 of 10 drug candidates were measured against SARS-CoV-2 in cell culture. Nilotinib and bemcentinib had EC50 values of 2.6 and 1.1 μM, respectively. In summary, the results of our computer-aided drug design provide a roadmap for rational drug design of Mpro inhibitors and the discovery of certified medications as COVID-19 antiviral therapeutics.
中文翻译:

重新确定尼洛替尼作为潜在 SARS-CoV-2 主要蛋白酶抑制剂的药物用途:来自计算和体外研究的见解
COVID-19 是一种急性病毒性肺炎,已成为一种毁灭性的流行病。药物再利用使研究人员能够找到 FDA 批准或研究药物的不同适应症。在当前的这项研究中,针对 COVID-19 M pro (PDB: 6LU7) 的一系列基于药效团和分子模型的筛选表明,药物银行数据库中的一个药物子集可能具有抗病毒活性。对来自药物银行的 8823 个最有希望的虚拟命中中的总共 44 个进行了分子动力学模拟实验,以探索它们与 SARS-CoV-2 M前活性位点相互作用的强度。MD 的研究结果表明,三种药物(DB04020、DB12411 和 DB11779)对 SARS-CoV-2 M pro的相对自由能非常低,并且在 His41 和 Met49 处相互作用。MD 模拟发现了与 Glu166 的额外相互作用,显着增强了结合亲和力。因此,Glu166 可能是基于结构的药物设计的一个有趣的目标。对基于分子对接的虚拟筛选的 44 个最有希望的命中进行了定量结构-活性关系分析。偏最小二乘回归以令人印象深刻的高精度准确预测了独立候选药物的结合能值。最后,在细胞培养物中测量了 10 种候选药物针对 SARS-CoV-2 的EC 50和 CC 50 。Nilotinib 和 bemcentinib 的 EC 50值分别为 2.6 和 1.1 μM。总之,我们的计算机辅助药物设计的结果为 M pro抑制剂的合理药物设计和发现经认证的 COVID-19 抗病毒治疗药物提供了路线图。
更新日期:2021-11-22
中文翻译:
重新确定尼洛替尼作为潜在 SARS-CoV-2 主要蛋白酶抑制剂的药物用途:来自计算和体外研究的见解
COVID-19 是一种急性病毒性肺炎,已成为一种毁灭性的流行病。药物再利用使研究人员能够找到 FDA 批准或研究药物的不同适应症。在当前的这项研究中,针对 COVID-19 M pro (PDB: 6LU7) 的一系列基于药效团和分子模型的筛选表明,药物银行数据库中的一个药物子集可能具有抗病毒活性。对来自药物银行的 8823 个最有希望的虚拟命中中的总共 44 个进行了分子动力学模拟实验,以探索它们与 SARS-CoV-2 M前活性位点相互作用的强度。MD 的研究结果表明,三种药物(DB04020、DB12411 和 DB11779)对 SARS-CoV-2 M pro的相对自由能非常低,并且在 His41 和 Met49 处相互作用。MD 模拟发现了与 Glu166 的额外相互作用,显着增强了结合亲和力。因此,Glu166 可能是基于结构的药物设计的一个有趣的目标。对基于分子对接的虚拟筛选的 44 个最有希望的命中进行了定量结构-活性关系分析。偏最小二乘回归以令人印象深刻的高精度准确预测了独立候选药物的结合能值。最后,在细胞培养物中测量了 10 种候选药物针对 SARS-CoV-2 的EC 50和 CC 50 。Nilotinib 和 bemcentinib 的 EC 50值分别为 2.6 和 1.1 μM。总之,我们的计算机辅助药物设计的结果为 M pro抑制剂的合理药物设计和发现经认证的 COVID-19 抗病毒治疗药物提供了路线图。










































 京公网安备 11010802027423号
京公网安备 11010802027423号