当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Enhanced Molecular Dynamics Method to Efficiently Increase the Discrimination Capability of Computational Protein–Protein Docking
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-10-15 , DOI: 10.1021/acs.jctc.1c00789 Nicola Scafuri 1 , Miguel A Soler 1 , Andrea Spitaleri 1 , Walter Rocchia 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-10-15 , DOI: 10.1021/acs.jctc.1c00789 Nicola Scafuri 1 , Miguel A Soler 1 , Andrea Spitaleri 1 , Walter Rocchia 1
Affiliation
|
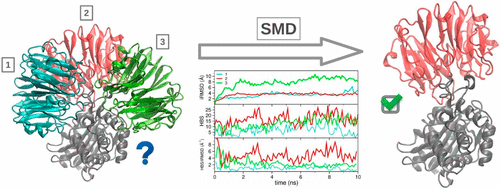
Protein–protein docking typically consists of the generation of putative binding conformations, which are subsequently ranked by fast heuristic scoring functions. The simplicity of these functions allows for computational efficiency but has severe repercussions on their discrimination capabilities. In this work, we show the effectiveness of suitable descriptors calculated along short scaled molecular dynamics runs in recognizing the nearest-native bound conformation among a set of putative structures generated by the HADDOCK tool for eight protein–protein systems.
中文翻译:

增强分子动力学方法有效提高计算蛋白质-蛋白质对接的辨别能力
蛋白质-蛋白质对接通常包括产生推定的结合构象,随后通过快速启发式评分函数对其进行排序。这些函数的简单性可以提高计算效率,但会对它们的辨别能力产生严重影响。在这项工作中,我们展示了沿着短尺度分子动力学运行计算的合适描述符在识别由 HADDOCK 工具为八种蛋白质-蛋白质系统生成的一组推定结构中的最近天然结合构象方面的有效性。
更新日期:2021-11-09
中文翻译:
增强分子动力学方法有效提高计算蛋白质-蛋白质对接的辨别能力
蛋白质-蛋白质对接通常包括产生推定的结合构象,随后通过快速启发式评分函数对其进行排序。这些函数的简单性可以提高计算效率,但会对它们的辨别能力产生严重影响。在这项工作中,我们展示了沿着短尺度分子动力学运行计算的合适描述符在识别由 HADDOCK 工具为八种蛋白质-蛋白质系统生成的一组推定结构中的最近天然结合构象方面的有效性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号