当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Prediction of ω-Transaminase Specificity by a Combination of Docking and Molecular Dynamics Simulations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-15 , DOI: 10.1021/acs.jcim.1c00617 Carlos Ramírez-Palacios 1, 2 , Hein J Wijma 1 , Sebastian Thallmair 2, 3 , Siewert J Marrink 2 , Dick B Janssen 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-15 , DOI: 10.1021/acs.jcim.1c00617 Carlos Ramírez-Palacios 1, 2 , Hein J Wijma 1 , Sebastian Thallmair 2, 3 , Siewert J Marrink 2 , Dick B Janssen 1
Affiliation
|
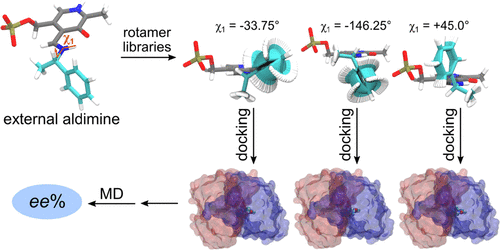
ω-Transaminases (ω-TAs) catalyze the conversion of ketones to chiral amines, often with high enantioselectivity and specificity, which makes them attractive for industrial production of chiral amines. Tailoring ω-TAs to accept non-natural substrates is necessary because of their limited substrate range. We present a computational protocol for predicting the enantioselectivity and catalytic selectivity of an ω-TA from Vibrio fluvialis with different substrates and benchmark it against 62 compounds gathered from the literature. Rosetta-generated complexes containing an external aldimine intermediate of the transamination reaction are used as starting conformations for multiple short independent molecular dynamics (MD) simulations. The combination of molecular docking and MD simulations ensures sufficient and accurate sampling of the relevant conformational space. Based on the frequency of near-attack conformations observed during the MD trajectories, enantioselectivities can be quantitatively predicted. The predicted enantioselectivities are in agreement with a benchmark dataset of experimentally determined ee% values. The substrate-range predictions can be based on the docking score of the external aldimine intermediate. The low computational cost required to run the presented framework makes it feasible for use in enzyme design to screen thousands of enzyme variants.
中文翻译:

通过对接和分子动力学模拟的组合计算预测 ω-转氨酶特异性
ω-转氨酶(ω-TAs)催化酮向手性胺的转化,通常具有高对映选择性和特异性,这使得它们在手性胺的工业生产中具有吸引力。调整 ω-TA 以接受非天然底物是必要的,因为它们的底物范围有限。我们提出了一种计算方案,用于预测来自河流弧菌的 ω-TA 的对映选择性和催化选择性使用不同的底物,并将其与从文献中收集的 62 种化合物进行对比。Rosetta 生成的复合物包含转氨反应的外部醛亚胺中间体,用作多个短独立分子动力学 (MD) 模拟的起始构象。分子对接和 MD 模拟的结合确保了相关构象空间的充分和准确采样。根据在 MD 轨迹期间观察到的近攻击构象的频率,可以定量预测对映选择性。预测的对映选择性与实验确定的ee的基准数据集一致% 值。底物范围预测可以基于外部醛亚胺中间体的对接分数。运行所提出的框架所需的低计算成本使其可用于酶设计以筛选数千种酶变体。
更新日期:2021-11-22
中文翻译:
通过对接和分子动力学模拟的组合计算预测 ω-转氨酶特异性
ω-转氨酶(ω-TAs)催化酮向手性胺的转化,通常具有高对映选择性和特异性,这使得它们在手性胺的工业生产中具有吸引力。调整 ω-TA 以接受非天然底物是必要的,因为它们的底物范围有限。我们提出了一种计算方案,用于预测来自河流弧菌的 ω-TA 的对映选择性和催化选择性使用不同的底物,并将其与从文献中收集的 62 种化合物进行对比。Rosetta 生成的复合物包含转氨反应的外部醛亚胺中间体,用作多个短独立分子动力学 (MD) 模拟的起始构象。分子对接和 MD 模拟的结合确保了相关构象空间的充分和准确采样。根据在 MD 轨迹期间观察到的近攻击构象的频率,可以定量预测对映选择性。预测的对映选择性与实验确定的ee的基准数据集一致% 值。底物范围预测可以基于外部醛亚胺中间体的对接分数。运行所提出的框架所需的低计算成本使其可用于酶设计以筛选数千种酶变体。










































 京公网安备 11010802027423号
京公网安备 11010802027423号