当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Improving Structure-Based Virtual Screening with Ensemble Docking and Machine Learning
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-15 , DOI: 10.1021/acs.jcim.1c00511 Joel Ricci-Lopez 1, 2 , Sergio A Aguila 2 , Michael K Gilson 3 , Carlos A Brizuela 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-15 , DOI: 10.1021/acs.jcim.1c00511 Joel Ricci-Lopez 1, 2 , Sergio A Aguila 2 , Michael K Gilson 3 , Carlos A Brizuela 1
Affiliation
|
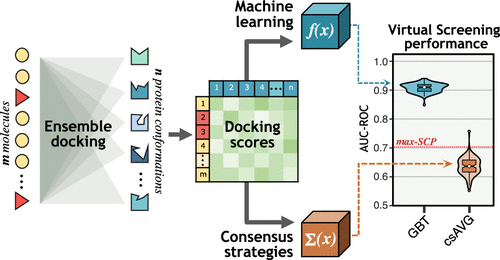
One of the main challenges of structure-based virtual screening (SBVS) is the incorporation of the receptor’s flexibility, as its explicit representation in every docking run implies a high computational cost. Therefore, a common alternative to include the receptor’s flexibility is the approach known as ensemble docking. Ensemble docking consists of using a set of receptor conformations and performing the docking assays over each of them. However, there is still no agreement on how to combine the ensemble docking results to obtain the final ligand ranking. A common choice is to use consensus strategies to aggregate the ensemble docking scores, but these strategies exhibit slight improvement regarding the single-structure approach. Here, we claim that using machine learning (ML) methodologies over the ensemble docking results could improve the predictive power of SBVS. To test this hypothesis, four proteins were selected as study cases: CDK2, FXa, EGFR, and HSP90. Protein conformational ensembles were built from crystallographic structures, whereas the evaluated compound library comprised up to three benchmarking data sets (DUD, DEKOIS 2.0, and CSAR-2012) and cocrystallized molecules. Ensemble docking results were processed through 30 repetitions of 4-fold cross-validation to train and validate two ML classifiers: logistic regression and gradient boosting trees. Our results indicate that the ML classifiers significantly outperform traditional consensus strategies and even the best performance case achieved with single-structure docking. We provide statistical evidence that supports the effectiveness of ML to improve the ensemble docking performance.
中文翻译:

通过集成对接和机器学习改进基于结构的虚拟筛选
基于结构的虚拟筛选 (SBVS) 的主要挑战之一是结合受体的灵活性,因为它在每次对接运行中的明确表示意味着高计算成本。因此,包含受体灵活性的常见替代方法是称为集成对接的方法。整体对接包括使用一组受体构象并对它们中的每一个进行对接分析。然而,对于如何结合整体对接结果来获得最终的配体排序,目前还没有达成一致。一个常见的选择是使用共识策略来聚合整体对接分数,但这些策略相对于单一结构方法略有改进。这里,我们声称,在整体对接结果上使用机器学习 (ML) 方法可以提高 SBVS 的预测能力。为了检验这一假设,选择了四种蛋白质作为研究案例:CDK2、FXa、EGFR 和 HSP90。蛋白质构象集合是从晶体结构构建的,而评估的化合物库包含多达三个基准数据集(DUD、DEKOIS 2.0 和 CSAR-2012)和共结晶分子。通过 30 次重复的 4 折交叉验证处理集成对接结果,以训练和验证两个 ML 分类器:逻辑回归和梯度提升树。我们的结果表明,ML 分类器明显优于传统的共识策略,甚至优于单结构对接实现的最佳性能案例。
更新日期:2021-11-22
中文翻译:
通过集成对接和机器学习改进基于结构的虚拟筛选
基于结构的虚拟筛选 (SBVS) 的主要挑战之一是结合受体的灵活性,因为它在每次对接运行中的明确表示意味着高计算成本。因此,包含受体灵活性的常见替代方法是称为集成对接的方法。整体对接包括使用一组受体构象并对它们中的每一个进行对接分析。然而,对于如何结合整体对接结果来获得最终的配体排序,目前还没有达成一致。一个常见的选择是使用共识策略来聚合整体对接分数,但这些策略相对于单一结构方法略有改进。这里,我们声称,在整体对接结果上使用机器学习 (ML) 方法可以提高 SBVS 的预测能力。为了检验这一假设,选择了四种蛋白质作为研究案例:CDK2、FXa、EGFR 和 HSP90。蛋白质构象集合是从晶体结构构建的,而评估的化合物库包含多达三个基准数据集(DUD、DEKOIS 2.0 和 CSAR-2012)和共结晶分子。通过 30 次重复的 4 折交叉验证处理集成对接结果,以训练和验证两个 ML 分类器:逻辑回归和梯度提升树。我们的结果表明,ML 分类器明显优于传统的共识策略,甚至优于单结构对接实现的最佳性能案例。










































 京公网安备 11010802027423号
京公网安备 11010802027423号