当前位置:
X-MOL 学术
›
ACS Macro Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Random Forest Predictor for Diblock Copolymer Phase Behavior
ACS Macro Letters ( IF 5.1 ) Pub Date : 2021-10-14 , DOI: 10.1021/acsmacrolett.1c00521 Akash Arora 1 , Tzyy-Shyang Lin 1 , Nathan J Rebello 1 , Sarah H M Av-Ron 1 , Hidenobu Mochigase 1 , Bradley D Olsen 1
ACS Macro Letters ( IF 5.1 ) Pub Date : 2021-10-14 , DOI: 10.1021/acsmacrolett.1c00521 Akash Arora 1 , Tzyy-Shyang Lin 1 , Nathan J Rebello 1 , Sarah H M Av-Ron 1 , Hidenobu Mochigase 1 , Bradley D Olsen 1
Affiliation
|
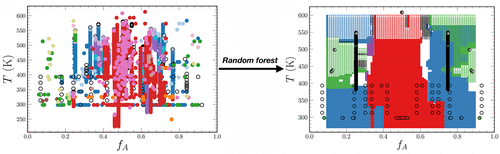
Physics-based models are the primary approach for modeling the phase behavior of block copolymers. However, the successful use of self-consistent field theory (SCFT) for designing new materials relies on the correct chemistry- and temperature-dependent Flory–Huggins interaction parameter χAB that quantifies the incompatibility between the two blocks A and B as well as accurate estimation of the ratio of Kuhn lengths (bA/bB) and block densities. This work uses machine learning to model the phase behavior of AB diblock copolymers by using the chemical identities of blocks directly, obviating the need for measurement of χAB and bA/bB. The random forest approach employed predicts the phase behavior with almost 90% accuracy after training on a data set of 4768 data points, almost twice the accuracy obtained using SCFT employing χAB from group contribution theory. The machine-learning model is notably sensitive toward the uncertainty in measuring molecular parameters; however, its accuracy still remains at least 60% even for highly uncertain experimental measurements. Accuracy is substantially reduced when extrapolating to chemistries outside the training set. This work demonstrates that a random forest phase predictor performs remarkably well in many scenarios, providing an opportunity to predict self-assembly without measurement of molecular parameters.
中文翻译:

二嵌段共聚物相行为的随机森林预测器
基于物理的模型是模拟嵌段共聚物相行为的主要方法。然而,成功使用自洽场论 ( SCFT ) 设计新材料依赖于正确的化学和温度相关的 Flory-Huggins 相互作用参数估计库恩长度(b A / b B)和块密度的比率。这项工作使用机器学习通过直接使用嵌段的化学特性来模拟 AB 二嵌段共聚物的相行为,无需测量 χ AB和b A / b B. 在对 4768 个数据点的数据集进行训练后,所采用的随机森林方法以几乎 90% 的准确度预测相位行为,几乎是使用来自群贡献理论的χ AB的SCFT 获得的准确度的两倍。机器学习模型对测量分子参数的不确定性特别敏感;然而,即使对于高度不确定的实验测量,其准确度仍保持至少 60%。当外推到训练集之外的化学物质时,准确性会大大降低。这项工作表明随机森林相位预测器在许多情况下都表现得非常好,提供了在不测量分子参数的情况下预测自组装的机会。
更新日期:2021-11-16
中文翻译:
二嵌段共聚物相行为的随机森林预测器
基于物理的模型是模拟嵌段共聚物相行为的主要方法。然而,成功使用自洽场论 ( SCFT ) 设计新材料依赖于正确的化学和温度相关的 Flory-Huggins 相互作用参数估计库恩长度(b A / b B)和块密度的比率。这项工作使用机器学习通过直接使用嵌段的化学特性来模拟 AB 二嵌段共聚物的相行为,无需测量 χ AB和b A / b B. 在对 4768 个数据点的数据集进行训练后,所采用的随机森林方法以几乎 90% 的准确度预测相位行为,几乎是使用来自群贡献理论的χ AB的SCFT 获得的准确度的两倍。机器学习模型对测量分子参数的不确定性特别敏感;然而,即使对于高度不确定的实验测量,其准确度仍保持至少 60%。当外推到训练集之外的化学物质时,准确性会大大降低。这项工作表明随机森林相位预测器在许多情况下都表现得非常好,提供了在不测量分子参数的情况下预测自组装的机会。










































 京公网安备 11010802027423号
京公网安备 11010802027423号