Frontiers of Chemical Science and Engineering ( IF 4.3 ) Pub Date : 2021-10-13 , DOI: 10.1007/s11705-021-2083-5 Yiming Ma 1, 2 , Zhenguo Gao 1, 2 , Peng Shi 1, 2 , Mingyang Chen 1, 2 , Songgu Wu 1, 2 , Jingkang Wang 1, 2 , Junbo Gong 1, 2 , Chao Yang 3 , Jingcai Cheng 3
|
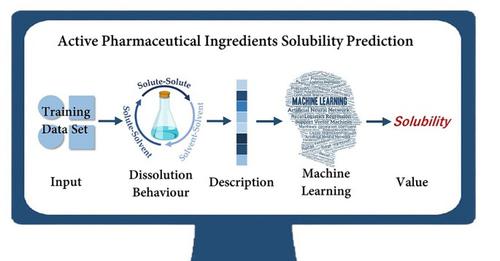
Solubility has been widely regarded as a fundamental property of small molecule drugs and drug candidates, as it has a profound impact on the crystallization process. Solubility prediction, as an alternative to experiments which can reduce waste and improve crystallization process efficiency, has attracted increasing attention. However, there are still many urgent challenges thus far. Herein we used seven descriptors based on understanding dissolution behavior to establish two solubility prediction models by machine learning algorithms. The solubility data of 120 active pharmaceutical ingredients (APIs) in ethanol were considered in the prediction models, which were constructed by random decision forests and artificial neural network with optimized data structure and model accuracy. Furthermore, a comparison with traditional prediction methods including the modified solubility equation and the quantitative structure-property relationships model was carried out. The highest accuracy shown by the testing set proves that the ML models have the best solubility prediction ability. Multiple linear regression and stepwise regression were used to further investigate the critical factor in determining solubility value. The results revealed that the API properties and the solute-solvent interaction both provide a nonnegligible contribution to the solubility value.
中文翻译:
基于机器学习的工业结晶中活性药物成分的溶解度预测和方法学评价
溶解度已被广泛认为是小分子药物和候选药物的基本特性,因为它对结晶过程具有深远的影响。溶解度预测作为可以减少浪费和提高结晶过程效率的实验的替代方法,已引起越来越多的关注。然而,到目前为止,仍然存在许多紧迫的挑战。在这里,我们基于对溶出行为的理解使用了七个描述符,通过机器学习算法建立了两个溶解度预测模型。预测模型考虑了120种活性药物成分(API)在乙醇中的溶解度数据,由随机决策森林和人工神经网络构建,优化了数据结构和模型精度。此外,与传统的预测方法进行了比较,包括修正的溶解度方程和定量结构性能关系模型。测试集显示的最高准确度证明 ML 模型具有最佳的溶解度预测能力。多元线性回归和逐步回归被用来进一步研究决定溶解度值的关键因素。结果表明,API 特性和溶质-溶剂相互作用对溶解度值的贡献都不可忽视。多元线性回归和逐步回归被用来进一步研究决定溶解度值的关键因素。结果表明,API 特性和溶质-溶剂相互作用对溶解度值的贡献都不可忽视。多元线性回归和逐步回归被用来进一步研究决定溶解度值的关键因素。结果表明,API 特性和溶质-溶剂相互作用对溶解度值的贡献都不可忽视。









































 京公网安备 11010802027423号
京公网安备 11010802027423号