当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crystallography and QM/MM Simulations Identify Preferential Binding of Hydrolyzed Carbapenem and Penem Antibiotics to the L1 Metallo-β-Lactamase in the Imine Form
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-12 , DOI: 10.1021/acs.jcim.1c00663 Rebecca M Twidale 1 , Philip Hinchliffe 2 , James Spencer 2 , Adrian J Mulholland 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-10-12 , DOI: 10.1021/acs.jcim.1c00663 Rebecca M Twidale 1 , Philip Hinchliffe 2 , James Spencer 2 , Adrian J Mulholland 1
Affiliation
|
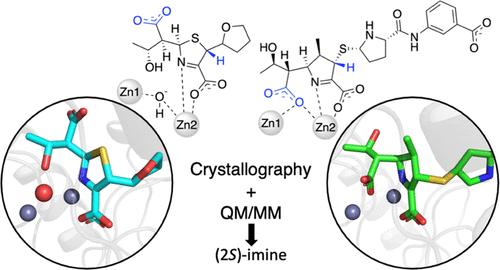
Widespread bacterial resistance to carbapenem antibiotics is an increasing global health concern. Resistance has emerged due to carbapenem-hydrolyzing enzymes, including metallo-β-lactamases (MβLs), but despite their prevalence and clinical importance, MβL mechanisms are still not fully understood. Carbapenem hydrolysis by MβLs can yield alternative product tautomers with the potential to access different binding modes. Here, we show that a combined approach employing crystallography and quantum mechanics/molecular mechanics (QM/MM) simulations allow tautomer assignment in MβL:hydrolyzed antibiotic complexes. Molecular simulations also examine (meta)stable species of alternative protonation and tautomeric states, providing mechanistic insights into β-lactam hydrolysis. We report the crystal structure of the hydrolyzed carbapenem ertapenem bound to the L1 MβL from Stenotrophomonas maltophilia and model alternative tautomeric and protonation states of both hydrolyzed ertapenem and faropenem (a related penem antibiotic), which display different binding modes with L1. We show how the structures of both complexed β-lactams are best described as the (2S)-imine tautomer with the carboxylate formed after β-lactam ring cleavage deprotonated. Simulations show that enamine tautomer complexes are significantly less stable (e.g., showing partial loss of interactions with the L1 binuclear zinc center) and not consistent with experimental data. Strong interactions of Tyr32 and one zinc ion (Zn1) with ertapenem prevent a C6 group rotation, explaining the different binding modes of the two β-lactams. Our findings establish the relative stability of different hydrolyzed (carba)penem forms in the L1 active site and identify interactions important to stable complex formation, information that should assist inhibitor design for this important antibiotic resistance determinant.
中文翻译:

晶体学和 QM/MM 模拟确定水解碳青霉烯和青霉烯抗生素与亚胺形式的 L1 金属-β-内酰胺酶的优先结合
细菌对碳青霉烯类抗生素的广泛耐药性是一个日益严重的全球健康问题。由于碳青霉烯类水解酶,包括金属-β-内酰胺酶 (MβLs),出现了耐药性,但尽管它们普遍存在且具有临床重要性,但 MβL 机制仍未完全了解。通过 MβLs 水解碳青霉烯可以产生替代产物互变异构体,有可能获得不同的结合模式。在这里,我们展示了采用晶体学和量子力学/分子力学 (QM/MM) 模拟的组合方法允许 MβL 中的互变异构体分配:水解抗生素复合物。分子模拟还检查了替代质子化和互变异构状态的(亚)稳定物质,提供了对 β-内酰胺水解的机理见解。嗜麦芽窄食单胞菌和水解厄他培南和法罗培南(一种相关的青霉烯类抗生素)的替代互变异构和质子化状态模型,它们与 L1 显示不同的结合模式。我们展示了如何最好地将两种复合 β-内酰胺的结构描述为 (2 S)-亚胺互变异构体与β-内酰胺环裂解去质子化后形成的羧酸盐。模拟表明,烯胺互变异构体复合物的稳定性明显降低(例如,显示与 L1 双核锌中心的相互作用部分丧失)并且与实验数据不一致。Tyr32 和一个锌离子 (Zn1) 与厄他培南的强相互作用阻止了 C6 基团的旋转,这解释了两种 β-内酰胺的不同结合模式。我们的研究结果确定了 L1 活性位点中不同水解 (carba)penem 形式的相对稳定性,并确定了对稳定复合物形成很重要的相互作用,这些信息应有助于针对这一重要的抗生素耐药性决定因素设计抑制剂。
更新日期:2021-10-12
中文翻译:
晶体学和 QM/MM 模拟确定水解碳青霉烯和青霉烯抗生素与亚胺形式的 L1 金属-β-内酰胺酶的优先结合
细菌对碳青霉烯类抗生素的广泛耐药性是一个日益严重的全球健康问题。由于碳青霉烯类水解酶,包括金属-β-内酰胺酶 (MβLs),出现了耐药性,但尽管它们普遍存在且具有临床重要性,但 MβL 机制仍未完全了解。通过 MβLs 水解碳青霉烯可以产生替代产物互变异构体,有可能获得不同的结合模式。在这里,我们展示了采用晶体学和量子力学/分子力学 (QM/MM) 模拟的组合方法允许 MβL 中的互变异构体分配:水解抗生素复合物。分子模拟还检查了替代质子化和互变异构状态的(亚)稳定物质,提供了对 β-内酰胺水解的机理见解。嗜麦芽窄食单胞菌和水解厄他培南和法罗培南(一种相关的青霉烯类抗生素)的替代互变异构和质子化状态模型,它们与 L1 显示不同的结合模式。我们展示了如何最好地将两种复合 β-内酰胺的结构描述为 (2 S)-亚胺互变异构体与β-内酰胺环裂解去质子化后形成的羧酸盐。模拟表明,烯胺互变异构体复合物的稳定性明显降低(例如,显示与 L1 双核锌中心的相互作用部分丧失)并且与实验数据不一致。Tyr32 和一个锌离子 (Zn1) 与厄他培南的强相互作用阻止了 C6 基团的旋转,这解释了两种 β-内酰胺的不同结合模式。我们的研究结果确定了 L1 活性位点中不同水解 (carba)penem 形式的相对稳定性,并确定了对稳定复合物形成很重要的相互作用,这些信息应有助于针对这一重要的抗生素耐药性决定因素设计抑制剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号