当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Spectra Calculations Using an Optimized Quasi-Regular Gaussian Basis and the Collocation Method
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-10-12 , DOI: 10.1021/acs.jctc.1c00805 Shane W Flynn 1 , Vladimir A Mandelshtam 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-10-12 , DOI: 10.1021/acs.jctc.1c00805 Shane W Flynn 1 , Vladimir A Mandelshtam 1
Affiliation
|
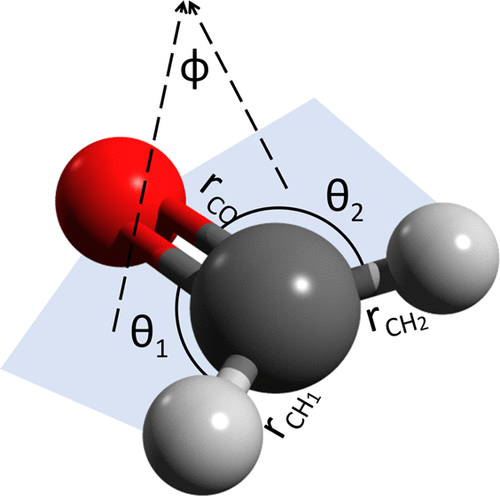
We revisit the collocation method of Manzhos and Carrington [ J. Chem. Phys., 2016, 145, 224110 ] in which a distributed localized (e.g., Gaussian) basis is used to set up a generalized eigenvalue problem to compute the eigenenergies and eigenfunctions of a molecular vibrational Hamiltonian. Although the resulting linear algebra problem involves full matrices, the method provides a number of important advantages, namely, (i) it is very simple both conceptually and numerically, (ii) it can be formulated using any set of internal molecular coordinates, (iii) it is flexible with respect to the choice of the basis, (iv) no integrals need to be computed, and (v) it has the potential to significantly reduce the basis size through optimizing the placement and the shapes of the basis functions. In the present paper, we explore the latter aspect of the method using the recently introduced, and here further improved, quasi-regular grids (QRGs). By computing the eigenenergies of the four-atom molecule of formaldehyde, we demonstrate that a QRG-based distributed Gaussian basis is superior to the previously used choices.
中文翻译:

使用优化的准正则高斯基和搭配方法计算分子光谱
我们重新审视 Manzhos 和 Carrington 的搭配方法 [ J. Chem. 物理。, 2016 , 145 ,224110 ] 其中分布式局部(例如,高斯)基用于建立广义特征值问题,以计算分子振动哈密顿量的特征能量和特征函数。虽然由此产生的线性代数问题涉及全矩阵,但该方法提供了许多重要的优点,即(i)它在概念上和数值上都非常简单,(ii)它可以使用任何一组内部分子坐标来公式化,(iii) ) 它在基的选择方面是灵活的,(iv) 不需要计算积分,并且 (v) 它有可能通过优化基函数的位置和形状来显着减小基大小。在本文中,我们使用最近引入的、这里进一步改进的准规则网格 (QRG) 来探索该方法的后一方面。
更新日期:2021-11-09
中文翻译:
使用优化的准正则高斯基和搭配方法计算分子光谱
我们重新审视 Manzhos 和 Carrington 的搭配方法 [ J. Chem. 物理。, 2016 , 145 ,










































 京公网安备 11010802027423号
京公网安备 11010802027423号